Alternative splicing (AS) of human telomerase catalytic subunit (hTERT, human telomerase reverse transcriptase) pre-mRNA strongly regulates telomerase activity. Several proteins can regulate AS in a cell type-specific manner and determine the functions of cells. In addition to being involved in telomerase activity regulation, AS provides cells with different splice variants that may have alternative biological activities. The modulation of telomerase activity through the induction of hTERT AS is involved in the development of different cancer types and embryos, and the differentiation of stem cells. Regulatory T cells may suppress the proliferation of target human and murine T and B lymphocytes and NK cells in a contact-independent manner involving activation of TERT AS.
- alternative splicing
- telomerase
- splice variants
- human telomerase reverse transcriptase (hTERT)
- telomeres
- lymphocytes
- endonuclease G
- apoptosis
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
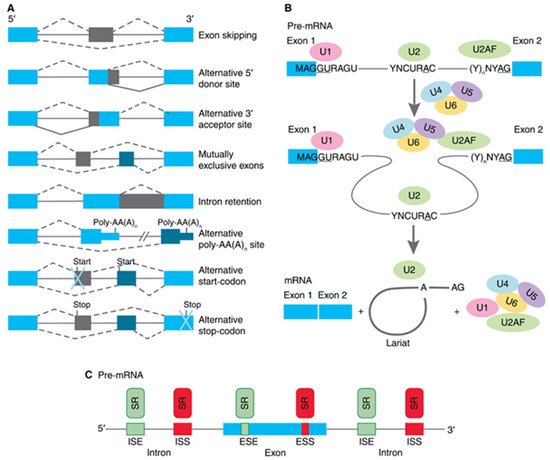
2. Regulatory Mechanisms for hTERT Pre-mRNA AS
2.1. Alternative Splice Variants of hTERT
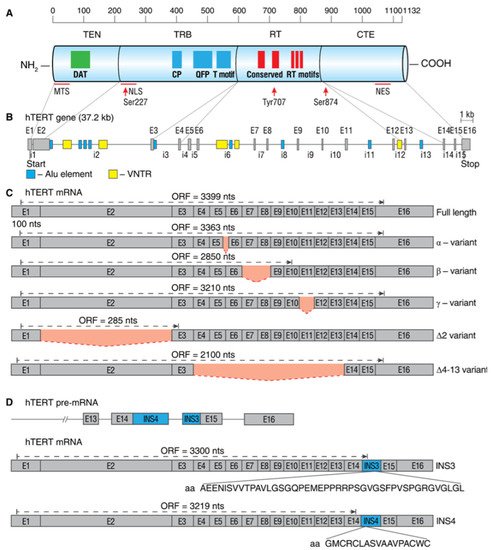
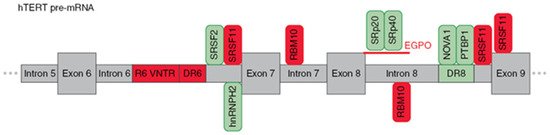
2.2. Regulation of hTERT Pre-mRNA AS
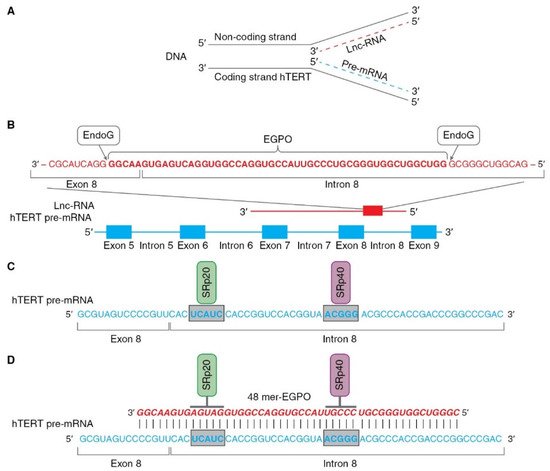
2.3. Modulation of hTERT Pre-mRNA AS by Endonuclease G
References
- Trybek, T.; Kowalik, A.; Góźdź, S.; Kowalska, A. Telomeres and telomerase in oncogenesis (review). Oncol. Lett. 2020, 20, 1015–1027.
- Ozturk, S. Telomerase activity and telomere length in male germ cells. Biol. Reprod. 2015, 92, 53.
- Hiyama, E.; Hiyama, K. Telomere and telomerase in stem cells. Br. J. Cancer 2007, 96, 1020–1024.
- Akbar, A.N.; Vukmanovic-Stejic, M. Telomerase in T lymphocytes: Use it and lose it? J. Immunol. 2007, 178, 6689–6694.
- Jakob, S.; Haendeler, J. Molecular mechanisms involved in endothelial cell aging: Role of telomerase reverse transcriptase. Z. Gerontol. Geriatr. 2007, 40, 334–338.
- Ishaq, A.; Hanson, P.S.; Morris, C.M.; Saretzki, G. Telomerase Activity is Downregulated Early during Human Brain Development. Genes 2016, 7, 27.
- Leão, R.; Apolónio, J.D.; Lee, D.; Figueiredo, A.; Tabori, U.; Castelo-Branco, P. Mechanisms of human telomerase reverse transcriptase (hTERT) regulation: Clinical impacts in cancer. J. Biomed. Sci. 2018, 25, 22.
- Liu, X.; Wang, Y.; Chang, G.; Wang, F.; Wang, F.; Geng, X. Alternative splicing of hTERT pre-mRNA: A potential strategy for the regulation of telomerase activity. Int. J. Mol. Sci. 2017, 18, 567.
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323.
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476.
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415.
- Miriami, E.; Sperling, R.; Sperling, J.; Motro, U. Regulation of splicing: The importance of being translatable. RNA 2004, 10, 1.
- Jurica, M.S.; Moore, M.J. Pre-mRNA splicing: Awash in a sea of proteins. Mol. Cell 2003, 12, 5–14.
- Savisaar, R.; Hurst, L.D. Exonic splice regulation imposes strong selection at synonymous sites. Genome Res. 2018, 28, 1442–1454.
- Sohail, M.; Xie, J. Diverse regulation of 3′ splice site usage. Cell. Mol. Life Sci. 2015, 72, 4771–4793.
- Brogna, S.; Wen, J. Nonsense-mediated mRNA decay (NMD) mechanisms. Nat. Struct. Mol. Biol. 2009, 16, 107–113.
- Faustino, N.A.; Cooper, T.A. Pre-mRNA splicing and human disease. Genes Dev. 2003, 17, 419–437.
- McKelvey, B.A.; Gilpatrick, T.; Wang, Y.; Timp, W.; Umbricht, C.B.; Zeiger, M.A. Characterization of Allele-Specific Regulation of Telomerase Reverse Transcriptase in Promoter Mutant Thyroid Cancer Cell Lines. Thyroid 2020, 30, 1470–1481.
- Wyatt, H.D.M.; West, S.C.; Beattie, T.L. InTERTpreting telomerase structure and function. Nucleic Acids Res. 2010, 38, 5609–5622.
- Zaug, A.J.; Podell, E.R.; Cech, T.R. Mutation in TERT separates processivity from anchor-site function. Nat. Struct. Mol. Biol. 2008, 15, 870–872.
- Jin, Y.; You, L.; Kim, H.J.; Lee, H.W. Telomerase reverse transcriptase contains a BH3-like motif and interacts with BCL-2 family members. Mol. Cells 2018, 41, 684–694.
- Xia, J.; Peng, Y.; Mian, I.S.; Lue, N.F. Identification of Functionally Important Domains in the N-Terminal Region of Telomerase Reverse Transcriptase. Mol. Cell. Biol. 2000, 20, 5196–5207.
- Bosoy, D.; Peng, Y.; Mian, I.S.; Lue, N.F. Conserved N-terminal motifs of telomerase reverse transcriptase required for ribonucleoprotein assembly in vivo. J. Biol. Chem. 2003, 278, 3882–3890.
- Lingner, J.; Hughes, T.R.; Shevchenko, A.; Mann, M.; Lundblad, V.; Cech, T.R. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science 1997, 276, 561–567.
- Huard, S.; Moriarty, T.J.; Autexier, C. The C terminus of the human telomerase reverse transcriptase is a determinant of enzyme processivity. Nucleic Acids Res. 2003, 31, 4059–4070.
- Li, H.; Zhao, L.; Yang, Z.; Funder, J.W.; Liu, J.P. Telomerase is controlled by protein kinase Cα in human breast cancer cells. J. Biol. Chem. 1998, 273, 33436–33442.
- Panneer Selvam, S.; De Palma, R.M.; Oaks, J.J.; Oleinik, N.; Peterson, Y.K.; Stahelin, R.V.; Skordalakes, E.; Ponnusamy, S.; Garrett-Mayer, E.; Smith, C.D.; et al. Binding of the sphingolipid S1P to hTERT stabilizes telomerase at the nuclear periphery by allosterically mimicking protein phosphorylation. Sci. Signal. 2015, 8, ra58.
- Jeong, S.A.; Kim, K.; Lee, J.H.; Cha, J.S.; Khadka, P.; Cho, H.S.; Chung, I.K. Akt-mediated phosphorylation increases the binding affinity of hTERT for importin α to promote nuclear translocation. J. Cell Sci. 2015, 128, 2287–2301.
- Chung, J.; Khadka, P.; Chung, I.K. Nuclear import of htert requires a bipartite nuclear localization signal and Akt-mediated phosphorylation. J. Cell Sci. 2012, 125, 2684–2697.
- Kang, S.S.; Kwon, T.; Kwon, D.Y.; Do, S.I. Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J. Biol. Chem. 1999, 274, 13085–13090.
- Büchner, N.; Zschauer, T.C.; Lukosz, M.; Altschmied, J.; Haendeler, J. Downregulation of mitochondrial telomerase reverse transcriptase induced by H2O2 is Src kinase dependent. Exp. Gerontol. 2010, 45, 558–562.
- Ludlow, A.T.; Slusher, A.L.; Sayed, M.E. Insights into telomerase/hTERT alternative splicing regulation using bioinformatics and network analysis in cancer. Cancers 2019, 11, 666.
- Zhu, S.; Rousseau, P.; Lauzon, C.; Gandin, V.; Topisirovic, I.; Autexier, C. Inactive C-terminal telomerase reverse transcriptase insertion splicing variants are dominant-negative inhibitors of telomerase. Biochimie 2014, 101, 93–103.
- Hrdlicková, R.; Nehyba, J.; Bose, H.R. Alternatively spliced telomerase reverse transcriptase variants lacking telomerase activity stimulate cell proliferation. Mol. Cell. Biol. 2012, 32, 4283–4296.
- Saebøe-Larssen, S.; Fossberg, E.; Gaudernack, G. Characterization of novel alternative splicing sites in human telomerase reverse transcriptase (hTERT): Analysis of expression and mutual correlation in mRNA isoforms from normal and tumour tissues. BMC Mol. Biol. 2006, 7, 26.
- Wong, M.S.; Wright, W.E.; Shay, J.W. Alternative splicing regulation of telomerase: A new paradigm? Trends Genet 2014, 30, 430–438.
- Listerman, I.; Sun, J.; Gazzaniga, F.S.; Lukas, J.L.; Blackburn, E.H. The major reverse transcriptase-incompetent splice variant of the human telomerase protein inhibits telomerase activity but protects from apoptosis. Cancer Res. 2013, 73, 2817–2828.
- Yi, X.; Shay, J.W.; Wright, W.E. Quantitation of telomerase components and hTERT mRNA splicing patterns in immortal human cells. Nucleic Acids Res. 2001, 29, 4818–4825.
- Withers, J.B.; Ashvetiya, T.; Beemon, K.L. Exclusion of Exon 2 Is a Common mRNA Splice Variant of Primate Telomerase Reverse Transcriptases. PLoS ONE 2012, 7, e48016.
- Zhdanov, D.D.; Vasina, D.A.; Orlova, V.S.; Gotovtseva, V.Y.; Bibikova, M.V.; Pokrovsky, V.S.; Pokrovskayaa, M.V.; Aleksandrova, S.S.; Sokolov, N.N. Apoptotic endonuclease EndoG induces alternative splicing of telomerase catalytic subunit hTERT and death of tumor cells. Biochem. Suppl. Ser. B Biomed. Chem. 2016, 10, 310–321.
- Yi, X.; White, D.M.; Aisner, D.L.; Baur, J.A.; Wright, W.E.; Shay, J.W. An alternate splicing variant of the human telomerase catalytic subunit inhibits telomerase activity. Neoplasia 2000, 2, 433–440.
- Zhdanov, D.D.; Vasina, D.A.; Grachev, V.A.; Orlova, E.V.; Orlova, V.S.; Pokrovskaya, M.V.; Alexandrova, S.S.; Sokolov, N.N. Alternative splicing of telomerase catalytic subunit hTERT generated by apoptotic endonuclease EndoG induces human CD4 + T cell death. Eur. J. Cell Biol. 2017, 96, 653–664.
- Zhdanov, D.D.; Vasina, D.A.; Orlova, E.V.; Orlova, V.S.; Pokrovskaya, M.V.; Aleksandrova, S.S.; Sokolov, N.N. Apoptotic endonuclease EndoG regulates alternative splicing of human telomerase catalytic subunit hTERT. Biochem. Suppl. Ser. B Biomed. Chem. 2017, 11, 154–165.
- Hisatomi, H.; Ohyashiki, K.; Ohyashiki, J.H.; Nagao, K.; Kanamaru, T.; Hirata, H.; Hibi, N.; Tsukada, Y. Expression profile of a gamma-deletion variant of the human telomerase reverse transcriptase gene. Neoplasia 2003, 5, 193–197.
- Kilian, A.; Bowtell, D.D.; Abud, H.E.; Hime, G.R.; Venter, D.J.; Keese, P.K.; Duncan, E.L.; Reddel, R.R.; Jefferson, R.A. Isolation of a candidate human telomerase catalytic subunit gene, which reveals complex splicing patterns in different cell types. Hum. Mol. Genet. 1997, 6, 2011–2019.
- Wright, W.E.; Piatyszek, M.A.; Rainey, W.E.; Byrd, W.; Shay, J.W. Telomerase activity in human germline and embryonic tissues and cells. Dev. Genet. 1996, 18, 173–179.
- Fleisig, H.B.; Hukezalie, K.R.; Thompson, C.A.H.; Au-Yeung, T.T.T.; Ludlow, A.T.; Zhao, C.R.; Wong, J.M.Y. Telomerase reverse transcriptase expression protects transformed human cells against DNA-damaging agents, and increases tolerance to chromosomal instability. Oncogene 2016, 35, 218–227.
- Cheng, D.; Zhao, Y.; Zhang, F.; Zhang, J.; Wang, S.; Zhu, J. Engineering a humanized telomerase reverse transcriptase gene in mouse embryonic stem cells. Sci. Rep. 2019, 9, 1–11.
- Tan, T.C.J.; Rahman, R.; Jaber-Hijazi, F.; Felix, D.A.; Chen, C.; Louis, E.J.; Aboobaker, A.; Haber, J.E. Telomere maintenance and telomerase activity are differentially regulated in asexual and sexual worms. Proc. Natl. Acad. Sci. USA 2012, 109, 4209–4214.
- Angelopoulou, K.; Zavlaris, M.; Papaioannou, N.; Vlemmas, I. Canis familiaris telomerase reverse transcriptase undergoes alternative splicing. Mamm. Genome 2008, 19, 647–653.
- Cieply, B.; Carstens, R.P. Functional roles of alternative splicing factors in human disease. Wiley Interdiscip. Rev. RNA 2015, 6, 311–326.
- Wong, M.S.; Chen, L.; Foster, C.; Kainthla, R.; Shay, J.W.; Wright, W.E. Regulation of telomerase alternative splicing: A target for chemotherapy. Cell Rep. 2013, 3, 1028–1035.
- Leem, S.H.; Londoño-Vallejo, J.A.; Kim, J.H.; Bui, H.; Tubacher, E.; Solomon, G.; Park, J.E.; Horikawa, I.; Kouprina, N.; Barrett, J.C.; et al. The human telomerase gene: Complete genomic sequence and analysis of tandem repeat polymorphisms in intronic regions. Oncogene 2002, 21, 769–777.
- Jin, Y.; Yang, Y.; Zhang, P. New insights into RNA secondary structure in the alternative splicing of pre-mRNAs. RNA Biol. 2011, 8, 450–457.
- Wong, M.S.; Shay, J.W.; Wright, W.E. Regulation of human telomerase splicing by RNA:RNA pairing. Nat. Commun. 2014, 5, 3306.
- Wu, J.Y.; Kar, A.; Kuo, D.; Yu, B.; Havlioglu, N. SRp54 (SFRS11), a Regulator for tau Exon 10 Alternative Splicing Identified by an Expression Cloning Strategy. Mol. Cell. Biol. 2006, 26, 6739–6747.
- Wang, F.; Cheng, Y.; Zhang, C.; Chang, G.; Geng, X. A novel antisense oligonucleotide anchored on the intronic splicing enhancer of hTERT pre-mRNA inhibits telomerase activity and induces apoptosis in glioma cells. J. Neurooncol. 2019, 143, 57–68.
- Xiao, W.; Chen, X.; Li, X.; Deng, K.; Liu, H.; Ma, J.; Wang, Z.; Hu, Y.; Hou, J. RBM10 regulates human TERT gene splicing and inhibits pancreatic cancer progression. Am. J. Cancer Res. 2021, 11, 157–170.
- Ludlow, A.T.; Wong, M.S.; Robin, J.D.; Batten, K.; Yuan, L.; Lai, T.P.; Dahlson, N.; Zhang, L.; Mender, I.; Tedone, E.; et al. NOVA1 regulates hTERT splicing and cell growth in non-small cell lung cancer. Nat. Commun. 2018, 9, 3112.
- Sayed, M.E.; Yuan, L.; Robin, J.D.; Tedone, E.; Batten, K.; Dahlson, N.; Wright, W.E.; Shay, J.W.; Ludlow, A.T. NOVA1 directs PTBP1 to hTERT pre-mRNA and promotes telomerase activity in cancer cells. Oncogene 2019, 38, 2937–2952.
- Villate, O.; Turatsinze, J.V.; Mascali, L.G.; Grieco, F.A.; Nogueira, T.C.; Cunha, D.A.; Nardelli, T.R.; Sammeth, M.; Salunkhe, V.A.; Esguerra, J.L.S.; et al. Nova1 is a master regulator of alternative splicing in pancreatic beta cells. Nucleic Acids Res. 2014, 42, 11818–11830.
- Zhdanov, D.D.; Plyasova, A.A.; Gladilina, Y.A.; Pokrovsky, V.S.; Grishin, D.V.; Grachev, V.A.; Orlova, V.S.; Pokrovskaya, M.V.; Alexandrova, S.S.; Lobaeva, T.A.; et al. Inhibition of telomerase activity by splice-switching oligonucleotides targeting the mRNA of the telomerase catalytic subunit affects proliferation of human CD4+ T lymphocytes. Biochem. Biophys. Res. Commun. 2019, 509, 790–796.
- Ito, T.; Watanabe, H.; Yamamichi, N.; Kondo, S.; Tando, T.; Haraguchi, T.; Mizutani, T.; Sakurai, K.; Fujita, S.; Izumi, T.; et al. Brm transactivates the telomerase reverse transcriptase (TERT) gene and modulates the splicing patterns of its transcripts in concert with p54 nrb. Biochem. J. 2008, 411, 201–209.
- Cerezo, A.; Kalthoff, H.; Schuermann, M.; Schäfer, B.; Boukamp, P. Dual regulation of telomerase activity through c-Myc-dependent inhibition and alternative splicing of hTERT. J. Cell Sci. 2002, 115, 1305–1312.
- Wang, F.; Chang, G.-M.; Geng, X.; Geng, X. Bioinformatics analysis of Exonic Splicing Enhancers (ESEs) for predicting potential regulatory elements of hTERT mRNA Splicing. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 526–536.
- Diener, T.; Neuhaus, M.; Koziel, R.; Micutkova, L.; Jansen-Dürr, P. Role of endonuclease G in senescence-associated cell death of human endothelial cells. Exp. Gerontol. 2010, 45, 638–644.
- Zhdanov, D.D.; Gladilina, Y.A.; Pokrovskaya, M.V.; Aleksandrova, S.S.; Grishin, D.V.; Podobed, O.V.; Sokolov, N.N. Induction of Alternative Splicing and Inhibition of Activity of Telomerase Catalytic Subunit by Apoptotic Endonuclease EndoG in Human, T., B, and NK Cells. Bull. Exp. Biol. Med. 2018, 164, 478–482.
- Ruiz-Carrillo, A.; Renaud, J. Endonuclease G: A (dG)n X (dC)n-specific DNase from higher eukaryotes. EMBO J. 1987, 6, 401–407.
- Li, L.Y.; Luo, X.; Wang, X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 2001, 412, 95–99.
- Zhdanov, D.D.; Fahmi, T.; Wang, X.; Apostolov, E.O.; Sokolov, N.N.; Javadov, S.; Basnakian, A.G. Regulation of Apoptotic Endonucleases by EndoG. DNA Cell Biol. 2015, 34, 316–326.
- Apostolov, E.O.; Wang, X.; Shah, S.V.; Basnakian, A.G. Role of EndoG in development and cell injury. Cell Death Differ. 2007, 14, 1971–1974.
- Zhdanov, D.D.; Pokrovsky, V.S.; Orlova, E.V.; Orlova, V.S.; Pokrovskaya, M.V.; Aleksandrova, S.S.; Sokolov, N.N. Intracellular localization of apoptotic endonuclease EndoG and splice-variants of telomerase catalytic subunit hTERT. Biochemistry 2017, 82, 894–905.
- Lee, J.S.; Seo, T.W.; Yi, J.H.; Shin, K.S.; Yoo, S.J. CHIP has a protective role against oxidative stress-induced cell death through specific regulation of endonuclease G. Cell Death Dis. 2013, 4, e666.
- Seo, T.W.; Lee, J.S.; Yoo, S.J. Cellular inhibitor of apoptosis protein 1 ubiquitinates endonuclease G but does not affect endonuclease G-mediated cell death. Biochem. Biophys. Res. Commun. 2014, 451, 644–649.
- Long, J.C.; Caceres, J.F. The SR protein family of splicing factors: Master regulators of gene expression. Biochem. J. 2009, 417, 15–27.
- Disterer, P.; Kryczka, A.; Liu, Y.; Badi, Y.E.; Wong, J.J.; Owen, J.S.; Khoo, B. Development of therapeutic splice-switching oligonucleotides. Hum. Gene Ther. 2014, 25, 587–598.
- Zhdanov, D.D.; Vasina, D.A.; Orlova, V.S.; Orlova, E.V.; Grishin, D.V.; Gladilina, Y.A.; Pokrovskaya, M.V.; Aleksandrova, S.S.; Sokolov, N.N. Induction of Apoptotic Endonuclease EndoG with DNA-Damaging Agents Initiates Alternative Splicing of Telomerase Catalytic Subunit hTERT and Inhibition of Telomerase Activity hTERT in Human CD4+ and CD8+T Lymphocytes. Biochem. Suppl. Ser. B Biomed. Chem. 2017, 63, 296–305.
- Zhdanov, D.D.; Gladilina, Y.A.; Orlova, V.S.; Grishin, D.V.; Pokrovskaya, M.V.; Aleksandrova, S.S.; Podobed, O.V.; Sokolov, N.N. Induction of Telomerase Catalytic Subunit Alternative Splicing by Apoptotic Endonuclease G in Mouse and Rat Lymphocytes. Cell Tissue Biol. 2018, 12, 104–115.
- Zhdanov, D.D.; Gladilina, Y.A.; Pokrovsky, V.S.; Grishin, D.V.; Grachev, V.A.; Orlova, V.S.; Pokrovskaya, M.V.; Alexandrova, S.S.; Plyasova, A.A.; Sokolov, N.N. Endonuclease G modulates the alternative splicing of deoxyribonuclease 1 mRNA in human CD4+ T lymphocytes and prevents the progression of apoptosis. Biochimie 2019, 157, 158–176.
- Zhdanov, D.D.; Gladilina, Y.A.; Grishin, D.V.; Pokrovsky, V.S.; Pokrovskaya, M.V.; Aleksandrova, S.S.; Sokolov, N.N. Apoptotic Endonuclease EndoG Induces Alternative Splicing of Telomerase TERT Catalytic Subunit, Caspase-2, DNase I, and BCL-x in Human, Murine, and Rat CD4+T Lymphocytes. Russ. J. Bioorg. Chem. 2018, 44, 90–103.