Aging is one of the hottest topics in biomedical research. Advances in research and medicine have helped to preserve human health, leading to an extension of life expectancy. However, the extension of life is an irreversible process accompanied by the development of aging-related conditions such as weakness, slower metabolism, and stiffness of vessels. It also debated that aging can be considered an actual disease with aging-derived comorbidities, including cancer or cardiovascular disease. Currently, cardiovascular disorders, including atherosclerosis, are considered premature aging and represent the first causes of death in developed countries, accounting for 31% of annual deaths globally. Emerging evidence has identified hypoxia-inducible factor-1α as a critical transcription factor with an essential role in aging-related pathology, in particular, regulating cellular senescence associated with cardiovascular aging.
- hypoxia-inducible factor-1α (HIF1α)
- vascular aging
- senescent cells
- endothelial cells
- vascular smooth muscle cells
- atherosclerosis
- extracellular vesicles
1. Hypoxia-Inducible Factors
| HIF Levels | Observations | Reference | |
|---|---|---|---|
| Pathology/Effect | Tissue/Cell Type | ||
| ↑ | Ischemic cardiovascular disease | Ischemic limb (ischemic calf muscle) (young WT mice) |
[54][22] |
| ↓ | Ischemic limb ischemic calf muscle (aging WT mice) |
||
| ↓ | Vascular remodeling | Femoral artery ligation in aging mice | [53][21] |
| ↓ | Femoral artery ligation in Hif1a+/− mice | ||
| ↑ (short-term) | Heart homeostasis | Heart | [52][19] |
| ↑ (long-term) | Cardiomyopathy | ||
| ↓ | Atherosclerosis (Premature aging disease) |
Pathogenesis of plaques (Promote angiogenesis, production of factor pro-atherosclerosis and recruitment of inflammatory cells) |
[3][20] |
| ↓ | Replicative senescence in vitro | Endothelial cells | [29][5] |
2. The Physio-Pathological Role of HIF in Aging
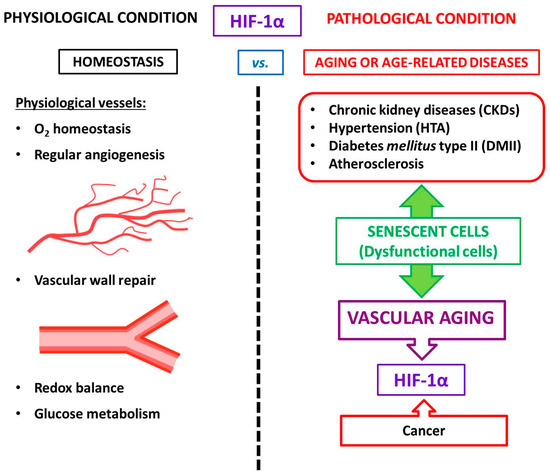
3. HIF-1α and Vascular Aging
4. HIF and Atherosclerosis
Although atherosclerosis has been considered chronic inflammation, intensive research in recent years has shown that it can also be considered an age-related pathology [28,100][71][72]. Many pieces of evidence have demonstrated the role of vascular senescence in atherogenesis [25,101][73][74]. We briefly mentioned above that senolytic drugs (anti-senescence) have been proposed as a therapeutic option for cellular aging and for treating human atherosclerosis [25,28][73][71]. However, although gerontologists have affirmed that atherosclerosis is associated with the characteristic features of aging in humans, cardiologists believe that aging is not a risk factor for atherosclerosis. This controversial subject was re-evaluated by Minamino et al., who demonstrated that senescent vascular cells accumulate in human atheroma and that vascular cells present features of dysfunction [102,103][75][76]. These and other findings suggest that cellular senescence contributes to atherosclerosis, which is a characteristic of aging in humans. As a model for premature aging disorder, atherosclerosis is the most common type of vascular aging where the cell vessels are susceptible to damage. Adding to the complex scenario for atherosclerosis, many studies suggest that ECs and VSMCs change and acquire features of senescent cells [104][77]. Moreover, during aging, blood vessels experience changes in compliance and release pro-inflammatory factors that promote atherosclerosis. Aging is associated with chronic low-grade inflammation that affects vascular and endothelial cells within the vascular wall during atherosclerosis. It is reasonable to believe that low-grade systemic inflammation may facilitate the senescent phenotype of ECs, which also contributes to the local inflammatory environment by SASP. These aging endothelium walls impair angiogenesis and decrease coagulation activity [104][77]. In an aged endothelium, senescent ECs failed to achieve HIF-1α stabilization and decreased miR-126 levels, which are both essential contributors to the maintenance of endothelium homeostasis [29][5]. In the vasculature, HIF-1α regulates pressure changes due to the negative regulation of TGF-β in ECs [85][56] (note that pressure overload leads to increased myocardial O2 consumption). In this regard, the anoxemia theory is defined as a condition of abnormal oxygenation of the arterial blood. This theory proposes that an imbalance between the demand for and supply of O2 in the arterial wall is a critical factor in the development of atherosclerosis [105][78]. As a consequence, macrophages become apoptotic, a necrotic core is built, and there is an eventual increase in angiogenesis, linking senescent cells to atherosclerosis progression [7][79]. Therefore, the anoxemia theory is postulated to explain the progress of atheroma plaque. In 2007, the presence of HIF-1α was described in atheroma plaque [106][80]. HIF-1α is a regulator of angiogenesis and inflammation in atherosclerotic plaque destabilization. Moreover, HIF-1α is associated with an increase in VEGF levels during the inflammatory process in atheroma plaque. Notably, activated macrophages in atherosclerosis have been observed to stabilize HIF-1α under normoxic conditions [106][80]. HIF-1α stabilization occurs due to the local relative hypoxia resulting from insufficient O2 diffusion in the thickened intima and increased O2 demand due to the local inflammatory response. If the O2 supply is restored, HIF-1α is degraded, which reduces VEGF production and subsequent angiogenic signaling [107][81]. Another study reported that HIF-1α increases as a consequence of neovascularization in complicated human atherosclerosis among human carotids, as well as in coronary plaques [105][78]. Mechanistically, the angiogenic effect of the alternatively spliced tissue factor (asTF) activates HIF-1/VEGF signaling [41][8]. Indeed, activated macrophages localized in atheroma plaques expressed HIF1-α and VEGF, confirming that both are critical to the regulation of human plaque angiogenesis and lesion progression. Therefore, HIF-1α mediates inflammation by promoting pro-inflammatory cytokine expression and, consequently, inflammatory cell recruitment [108,109][82][83]. In macrophages, HIF-1α regulates the expression of one of the major pro-inflammatory cytokines, IL-1β [110][84]. Pro-inflammatory and pro-angiogenic activities are induced in endothelial cells exposed to IL-1β stimulation [111][85]. Notably, anti-inflammatory IL-1β therapy led to a significantly lower rate of recurrent cardiovascular events [112][86]. Another IL-1 family member, IL-1α, is mainly associated with endothelial cell senescence and atherosclerosis [113][87]. Both IL-1α and IL-1β are minor components of SASP; however, these two cytokines are essential for boosting IL-6 and IL-8, which are secreted in large quantities by senescent endothelial cells [114,115][88][89] (Figure 2).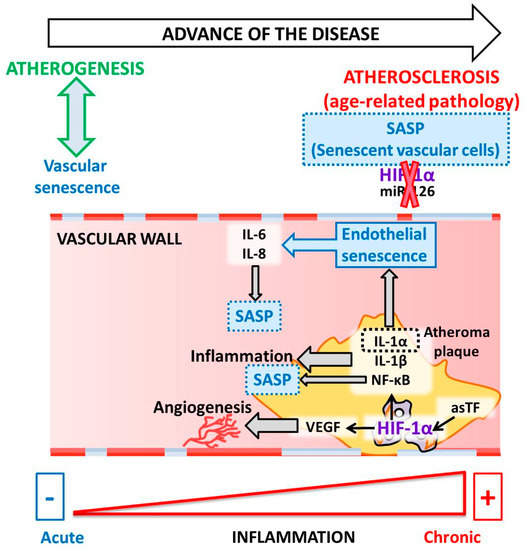
References
- López-Lázaro, M. HIF-1: Hypoxia-inducible factor or dysoxia-inducible factor? FASEB J. 2006, 20, 828–832.
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309.
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975.
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408.
- Alique, M.; Bodega, G.; Giannarelli, C.; Carracedo, J.; Ramírez, R. MicroRNA-126 regulates Hypoxia-Inducible Factor-1α which inhibited migration, proliferation, and angiogenesis in replicative endothelial senescence. Sci. Rep. 2019, 9, 7381.
- Rey, S.; Semenza, G.L. Hypoxia-inducible factor-1-dependent mechanisms of vascularization and vascular remodelling. Cardiovasc. Res. 2010, 86, 236–242.
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133.
- Giannarelli, C.; Alique, M.; Rodriguez, D.T.; Yang, D.K.; Jeong, D.; Calcagno, C.; Hutter, R.; Millon, A.; Kovacic, J.C.; Weber, T.; et al. Alternatively Spliced Tissue Factor Promotes Plaque Angiogenesis Through the Activation of Hypoxia-Inducible Factor-1 alpha and Vascular Endothelial Growth Factor Signaling. Circulation 2014, 130, 1274–1286.
- Elvidge, G.P.; Glenny, L.; Appelhoff, R.J.; Ratcliffe, P.J.; Ragoussis, J.; Gleadle, J.M. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition: The role of HIF-1alpha, HIF-2alpha, and other pathways. J. Biol. Chem. 2006, 281, 15215–15226.
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669.
- Lim, C.S.; Kiriakidis, S.; Sandison, A.; Paleolog, E.M.; Davies, A.H. Hypoxia-inducible factor pathway and diseases of the vascular wall. J. Vasc. Surg. 2013, 58, 219–230.
- Fernandez Esmerats, J.; Villa-Roel, N.; Kumar, S.; Gu, L.; Salim, M.T.; Ohh, M.; Taylor, W.R.; Nerem, R.M.; Yoganathan, A.P.; Jo, H. Disturbed Flow Increases UBE2C (Ubiquitin E2 Ligase C) via Loss of miR-483-3p, Inducing Aortic Valve Calcification by the pVHL (von Hippel-Lindau Protein) and HIF-1alpha (Hypoxia-Inducible Factor-1alpha) Pathway in Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 467–481.
- Wu, D.; Huang, R.T.; Hamanaka, R.B.; Krause, M.; Oh, M.J.; Kuo, C.H.; Nigdelioglu, R.; Meliton, A.Y.; Witt, L.; Dai, G.; et al. HIF-1alpha is required for disturbed flow-induced metabolic reprogramming in human and porcine vascular endothelium. Elife 2017, 6.
- Liu, D.; Lei, L.; Desir, M.; Huang, Y.; Cleman, J.; Jiang, W.; Fernandez-Hernando, C.; Di Lorenzo, A.; Sessa, W.C.; Giordano, F.J. Smooth Muscle Hypoxia-Inducible Factor 1alpha Links Intravascular Pressure and Atherosclerosis--Brief Report. Arterioscle. Thromb. Vasc. Biol. 2016, 36, 442–445.
- Wang, W.; Xu, B.; Xuan, H.; Ge, Y.; Wang, Y.; Wang, L.; Huang, J.; Fu, W.; Michie, S.A.; Dalman, R.L. Hypoxia-inducible factor 1 in clinical and experimental aortic aneurysm disease. J. Vasc. Surg. 2018, 68, 1538–1550.
- Imanishi, M.; Chiba, Y.; Tomita, N.; Matsunaga, S.; Nakagawa, T.; Ueno, M.; Yamamoto, K.; Tamaki, T.; Tomita, S. Hypoxia-Inducible Factor-1alpha in Smooth Muscle Cells Protects Against Aortic Aneurysms-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2158–2162.
- Semenza, G.L. HIF-1: Mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol. 2000, 88, 1474–1480.
- Abe, H.; Semba, H.; Takeda, N. The Roles of Hypoxia Signaling in the Pathogenesis of Cardiovascular Diseases. J. Atheroscler. Thromb. 2017, 24, 884–894.
- Hölscher, M.; Schäfer, K.; Krull, S.; Farhat, K.; Hesse, A.; Silter, M.; Lin, Y.; Pichler, B.J.; Thistlethwaite, P.; El-Armouche, A.; et al. Unfavourable consequences of chronic cardiac HIF-1α stabilization. Cardiovasc. Res. 2012, 94, 77–86.
- Kaluz, S.; Tan, C.; Van Meir, E.G. Taking a HIF pill for old age diseases? Aging 2018, 10, 290–292.
- Semenza, G.L. Hypoxia-inducible factor 1 and cardiovascular disease. Annu. Rev. Physiol. 2014, 76, 39–56.
- Bosch-Marce, M.; Okuyama, H.; Wesley, J.B.; Sarkar, K.; Kimura, H.; Liu, Y.V.; Zhang, H.; Strazza, M.; Rey, S.; Savino, L.; et al. Effects of aging and hypoxia-inducible factor-1 activity on angiogenic cell mobilization and recovery of perfusion after limb ischemia. Circ. Res. 2007, 101, 1310–1318.
- Bianchi, G.; Di Giulio, C.; Rapino, C.; Rapino, M.; Antonucci, A.; Cataldi, A. p53 and p66 proteins compete for hypoxia-inducible factor 1 alpha stabilization in young and old rat hearts exposed to intermittent hypoxia. Gerontology 2006, 52, 17–23.
- Rapino, C.; Bianchi, G.; Di Giulio, C.; Centurione, L.; Cacchio, M.; Antonucci, A.; Cataldi, A. HIF-1alpha cytoplasmic accumulation is associated with cell death in old rat cerebral cortex exposed to intermittent hypoxia. Aging Cell 2005, 4, 177–185.
- Mehta, R.; Steinkraus, K.A.; Sutphin, G.L.; Ramos, F.J.; Shamieh, L.S.; Huh, A.; Davis, C.; Chandler-Brown, D.; Kaeberlein, M. Proteasomal regulation of the hypoxic response modulates aging in C. elegans. Science 2009, 324, 1196–1198.
- Leiser, S.F.; Begun, A.; Kaeberlein, M. HIF-1 modulates longevity and healthspan in a temperature-dependent manner. Aging Cell 2011, 10, 318–326.
- Leiser, S.F.; Fletcher, M.; Begun, A.; Kaeberlein, M. Life-span extension from hypoxia in Caenorhabditis elegans requires both HIF-1 and DAF-16 and is antagonized by SKN-1. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 1135–1144.
- Leiser, S.F.; Kaeberlein, M. The hypoxia-inducible factor HIF-1 functions as both a positive and negative modulator of aging. Biol. Chem. 2010, 391, 1131–1137.
- Regina, C.; Panatta, E.; Candi, E.; Melino, G.; Amelio, I.; Balistreri, C.R.; Annicchiarico-Petruzzelli, M.; Di Daniele, N.; Ruvolo, G. Vascular ageing and endothelial cell senescence: Molecular mechanisms of physiology and diseases. Mech. Ageing Dev. 2016, 159, 14–21.
- Rezvani, H.R.; Ali, N.; Serrano-Sanchez, M.; Dubus, P.; Varon, C.; Ged, C.; Pain, C.; Cario-André, M.; Seneschal, J.; Taïeb, A.; et al. Loss of epidermal hypoxia-inducible factor-1α accelerates epidermal aging and affects re-epithelialization in human and mouse. J. Cell Sci. 2011, 124, 4172–4183.
- Ebersole, J.L.; Novak, M.J.; Orraca, L.; Martinez-Gonzalez, J.; Kirakodu, S.; Chen, K.C.; Stromberg, A.; Gonzalez, O.A. Hypoxia-inducible transcription factors, HIF1A and HIF2A, increase in aging mucosal tissues. Immunology 2018, 154, 452–464.
- Schmitt, R. Senotherapy: Growing old and staying young? Pflugers Arch. 2017, 469, 1051–1059.
- Gardner, S.E.; Humphry, M.; Bennett, M.R.; Clarke, M.C. Senescent Vascular Smooth Muscle Cells Drive Inflammation Through an Interleukin-1α-Dependent Senescence-Associated Secretory Phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1963–1974.
- Liu, Z.; Kuo, P.L.; Horvath, S.; Crimmins, E.; Ferrucci, L.; Levine, M. A new aging measure captures morbidity and mortality risk across diverse subpopulations from NHANES IV: A cohort study. PLoS Med. 2018, 15, e1002718.
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: Effects of telomerase and oxidative stress. Circ. Res. 2006, 99, 156–164.
- Morgan, R.G.; Ives, S.J.; Lesniewski, L.A.; Cawthon, R.M.; Andtbacka, R.H.; Noyes, R.D.; Richardson, R.S.; Donato, A.J. Age-related telomere uncapping is associated with cellular senescence and inflammation independent of telomere shortening in human arteries. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H251–H258.
- Ortiz-Montero, P.; Londoño-Vallejo, A.; Vernot, J.P. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun. Signal 2017, 15, 17.
- Young, A.P.; Schlisio, S.; Minamishima, Y.A.; Zhang, Q.; Li, L.; Grisanzio, C.; Signoretti, S.; Kaelin, W.G. VHL loss actuates a HIF-independent senescence programme mediated by Rb and p400. Nat. Cell Biol. 2008, 10, 361–369.
- Rossman, M.J.; Kaplon, R.E.; Hill, S.D.; McNamara, M.N.; Santos-Parker, J.R.; Pierce, G.L.; Seals, D.R.; Donato, A.J. Endothelial cell senescence with aging in healthy humans: Prevention by habitual exercise and relation to vascular endothelial function. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H890–H895.
- Montgomery, R.L.; van Rooij, E. MicroRNA regulation as a therapeutic strategy for cardiovascular disease. Curr. Drug Targets 2010, 11, 936–942.
- Small, E.M.; Olson, E.N. Pervasive roles of microRNAs in cardiovascular biology. Nature 2011, 469, 336–342.
- Hergenreider, E.; Heydt, S.; Tréguer, K.; Boettger, T.; Horrevoets, A.J.; Zeiher, A.M.; Scheffer, M.P.; Frangakis, A.S.; Yin, X.; Mayr, M.; et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat. Cell Biol. 2012, 14, 249–256.
- Zernecke, A.; Bidzhekov, K.; Noels, H.; Shagdarsuren, E.; Gan, L.; Denecke, B.; Hristov, M.; Köppel, T.; Jahantigh, M.N.; Lutgens, E.; et al. Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci. Signal 2009, 2, ra81.
- Fernández-Hernando, C.; Suárez, Y. MicroRNAs in endothelial cell homeostasis and vascular disease. Curr. Opin. Hematol. 2018, 25, 227–236.
- Van Solingen, C.; de Boer, H.C.; Bijkerk, R.; Monge, M.; van Oeveren-Rietdijk, A.M.; Seghers, L.; de Vries, M.R.; van der Veer, E.P.; Quax, P.H.; Rabelink, T.J.; et al. MicroRNA-126 modulates endothelial SDF-1 expression and mobilization of Sca-1+/Lin− progenitor cells in ischaemia. Cardiovasc. Res. 2011, 92, 449–455.
- Stellos, K.; Bigalke, B.; Langer, H.; Geisler, T.; Schad, A.; Kögel, A.; Pfaff, F.; Stakos, D.; Seizer, P.; Müller, I.; et al. Expression of stromal-cell-derived factor-1 on circulating platelets is increased in patients with acute coronary syndrome and correlates with the number of CD34+ progenitor cells. Eur. Heart J. 2009, 30, 584–593.
- Qu, M.; Pan, J.; Shi, X.; Zhang, Z.; Tang, Y.; Yang, G. MicroRNA-126 is a prospective target for vascular disease. Neuroimmunol. Neuroinflam. 2018, 5, 10.
- Zhou, J.; Li, Y.S.; Nguyen, P.; Wang, K.C.; Weiss, A.; Kuo, Y.C.; Chiu, J.J.; Shyy, J.Y.; Chien, S. Regulation of vascular smooth muscle cell turnover by endothelial cell-secreted microRNA-126: Role of shear stress. Circ. Res. 2013, 113, 40–51.
- Hao, X.Z.; Fan, H.M. Identification of miRNAs as atherosclerosis biomarkers and functional role of miR-126 in atherosclerosis progression through MAPK signalling pathway. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 2725–2733.
- Semenza, G.L. Pharmacologic Targeting of Hypoxia-Inducible Factors. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 379–403.
- Zhang, X.; Sarkar, K.; Rey, S.; Sebastian, R.; Andrikopoulou, E.; Marti, G.P.; Fox-Talbot, K.; Semenza, G.L.; Harmon, J.W. Aging impairs the mobilization and homing of bone marrow-derived angiogenic cells to burn wounds. J. Mol. Med. 2011, 89, 985–995.
- Nilsson, P.M.; Lurbe, E.; Laurent, S. The early life origins of vascular ageing and cardiovascular risk: The EVA syndrome. J. Hypertens. 2008, 26, 1049–1057.
- Dai, L.; Qureshi, A.R.; Witasp, A.; Lindholm, B.; Stenvinkel, P. Early Vascular Ageing and Cellular Senescence in Chronic Kidney Disease. Comput. Struct. Biotechnol. J. 2019, 17, 721–729.
- Shanahan, C.M. Mechanisms of vascular calcification in CKD-evidence for premature ageing? Nat. Rev. Nephrol. 2013, 9, 661–670.
- Shroff, R.; Long, D.A.; Shanahan, C. Mechanistic insights into vascular calcification in CKD. J. Am. Soc. Nephrol. 2013, 24, 179–189.
- Wei, H.; Bedja, D.; Koitabashi, N.; Xing, D.; Chen, J.; Fox-Talbot, K.; Rouf, R.; Chen, S.; Steenbergen, C.; Harmon, J.W.; et al. Endothelial expression of hypoxia-inducible factor 1 protects the murine heart and aorta from pressure overload by suppression of TGF-β signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E841–E850.
- Yeo, E.J. Hypoxia and aging. Exp. Mol. Med. 2019, 51, 67.
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013, 18, 416–430.
- Kume, S.; Uzu, T.; Horiike, K.; Chin-Kanasaki, M.; Isshiki, K.; Araki, S.; Sugimoto, T.; Haneda, M.; Kashiwagi, A.; Koya, D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J. Clin. Investig. 2010, 120, 1043–1055.
- Ryu, D.R.; Yu, M.R.; Kong, K.H.; Kim, H.; Kwon, S.H.; Jeon, J.S.; Han, D.C.; Noh, H. Sirt1-hypoxia-inducible factor-1α interaction is a key mediator of tubulointerstitial damage in the aged kidney. Aging Cell 2019, 18, e12904.
- Laemmle, A.; Lechleiter, A.; Roh, V.; Schwarz, C.; Portmann, S.; Furer, C.; Keogh, A.; Tschan, M.P.; Candinas, D.; Vorburger, S.A.; et al. Inhibition of SIRT1 impairs the accumulation and transcriptional activity of HIF-1α protein under hypoxic conditions. PLoS ONE 2012, 7, e33433.
- Yuan, Y.; Cruzat, V.F.; Newsholme, P.; Cheng, J.; Chen, Y.; Lu, Y. Regulation of SIRT1 in aging: Roles in mitochondrial function and biogenesis. Mech. Ageing Dev. 2016, 155, 10–21.
- Kitada, M.; Ogura, Y.; Koya, D. The protective role of Sirt1 in vascular tissue: Its relationship to vascular aging and atherosclerosis. Aging 2016, 8, 2290–2307.
- Orimo, M.; Minamino, T.; Miyauchi, H.; Tateno, K.; Okada, S.; Moriya, J.; Komuro, I. Protective role of SIRT1 in diabetic vascular dysfunction. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 889–894.
- Ota, H.; Eto, M.; Ogawa, S.; Iijima, K.; Akishita, M.; Ouchi, Y. SIRT1/eNOS axis as a potential target against vascular senescence, dysfunction and atherosclerosis. J. Atheroscler. Thromb. 2010, 17, 431–435.
- Zu, Y.; Liu, L.; Lee, M.Y.; Xu, C.; Liang, Y.; Man, R.Y.; Vanhoutte, P.M.; Wang, Y. SIRT1 promotes proliferation and prevents senescence through targeting LKB1 in primary porcine aortic endothelial cells. Circ. Res. 2010, 106, 1384–1393.
- Rivard, A.; Berthou-Soulie, L.; Principe, N.; Kearney, M.; Curry, C.; Branellec, D.; Semenza, G.L.; Isner, J.M. Age-dependent defect in vascular endothelial growth factor expression is associated with reduced hypoxia-inducible factor 1 activity. J. Biol. Chem. 2000, 275, 29643–29647.
- Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998, 352, 837–853.
- Macciò, A.; Madeddu, C. Management of anemia of inflammation in the elderly. Anemia 2012, 2012, 563251.
- Nakayama, T.; Kurobe, H.; Sugasawa, N.; Kinoshita, H.; Higashida, M.; Matsuoka, Y.; Yoshida, Y.; Hirata, Y.; Sakata, M.; Maxfield, M.W.; et al. Role of macrophage-derived hypoxia-inducible factor (HIF)-1α as a mediator of vascular remodelling. Cardiovasc. Res. 2013, 99, 705–715.
- Minamino, T.; Komuro, I. Vascular cell senescence: Contribution to atherosclerosis. Circ. Res. 2007, 100, 15–26.
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695.
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016, 15, 973–977.
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477.
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544.
- Minamino, T.; Yoshida, T.; Tateno, K.; Miyauchi, H.; Zou, Y.; Toko, H.; Komuro, I. Ras induces vascular smooth muscle cell senescence and inflammation in human atherosclerosis. Circulation 2003, 108, 2264–2269.
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Tateno, K.; Kunieda, T.; Komuro, I. Vascular cell senescence and vascular aging. J. Mol. Cell Cardiol. 2004, 36, 175–183.
- Ferns, G.A.A.; Heikal, L. Hypoxia in Atherogenesis. Angiology 2017, 68, 472–493.
- Alique, M.; Ramírez-Carracedo, R.; Bodega, G.; Carracedo, J.; Ramírez, R. Senescent Microvesicles: A Novel Advance in Molecular Mechanisms of Atherosclerotic Calcification. Int. J. Mol. Sci. 2018, 19.
- Vink, A.; Schoneveld, A.H.; Lamers, D.; Houben, A.J.; van der Groep, P.; van Diest, P.J.; Pasterkamp, G. HIF-1 alpha expression is associated with an atheromatous inflammatory plaque phenotype and upregulated in activated macrophages. Atherosclerosis 2007, 195, e69–e75.
- Camaré, C.; Pucelle, M.; Nègre-Salvayre, A.; Salvayre, R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017, 12, 18–34.
- Parma, L.; Baganha, F.; Quax, P.H.A.; de Vries, M.R. Plaque angiogenesis and intraplaque hemorrhage in atherosclerosis. Eur. J. Pharmacol. 2017, 816, 107–115.
- Ramkhelawon, B.; Yang, Y.; van Gils, J.M.; Hewing, B.; Rayner, K.J.; Parathath, S.; Guo, L.; Oldebeken, S.; Feig, J.L.; Fisher, E.A.; et al. Hypoxia induces netrin-1 and Unc5b in atherosclerotic plaques: Mechanism for macrophage retention and survival. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1180–1188.
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242.
- Mohr, T.; Haudek-Prinz, V.; Slany, A.; Grillari, J.; Micksche, M.; Gerner, C. Proteome profiling in IL-1beta and VEGF-activated human umbilical vein endothelial cells delineates the interlink between inflammation and angiogenesis. PLoS ONE 2017, 12, e0179065.
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131.
- Mariotti, M.; Castiglioni, S.; Bernardini, D.; Maier, J.A. Interleukin 1 alpha is a marker of endothelial cellular senescent. Immun. Ageing 2006, 3, 4.
- Apte, R.N.; Dotan, S.; Elkabets, M.; White, M.R.; Reich, E.; Carmi, Y.; Song, X.; Dvozkin, T.; Krelin, Y.; Voronov, E. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis. Rev. 2006, 25, 387–408.
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. USA 2009, 106, 17031–17036.
- Zouein, F.A.; Booz, G.W.; Altara, R. STAT3 and Endothelial Cell-Cardiomyocyte Dialog in Cardiac Remodeling. Front. Cardiovasc. Med. 2019, 6, 50.
- Chen, Q.; Lv, J.; Yang, W.; Xu, B.; Wang, Z.; Yu, Z.; Wu, J.; Yang, Y.; Han, Y. Targeted inhibition of STAT3 as a potential treatment strategy for atherosclerosis. Theranostics 2019, 9, 6424–6442.
- Ribeiro, S.; Belo, L.; Reis, F.; Santos-Silva, A. The HIF System Response to ESA Therapy in CKD-Anemia. In Hypoxia and Human Diseases; InTech Open: London, UK, 2017.
- Cao, L.; Li, W.; Kim, S.; Brodie, S.G.; Deng, C.X. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev. 2003, 17, 201–213.
- Lacolley, P.; Regnault, V.; Avolio, A.P. Smooth muscle cell and arterial aging: Basic and clinical aspects. Cardiovasc. Res. 2018, 114, 513–528.
- Akhtar, S.; Hartmann, P.; Karshovska, E.; Rinderknecht, F.A.; Subramanian, P.; Gremse, F.; Grommes, J.; Jacobs, M.; Kiessling, F.; Weber, C.; et al. Endothelial Hypoxia-Inducible Factor-1α Promotes Atherosclerosis and Monocyte Recruitment by Upregulating MicroRNA-19a. Hypertension 2015, 66, 1220–1226.
- Aarup, A.; Pedersen, T.X.; Junker, N.; Christoffersen, C.; Bartels, E.D.; Madsen, M.; Nielsen, C.H.; Nielsen, L.B. Hypoxia-Inducible Factor-1α Expression in Macrophages Promotes Development of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1782–1790.
- Sage, A.P.; Tintut, Y.; Demer, L.L. Regulatory mechanisms in vascular calcification. Nat. Rev. Cardiol. 2010, 7, 528–536.
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600.
- Pescatore, L.A.; Gamarra, L.F.; Liberman, M. Multifaceted Mechanisms of Vascular Calcification in Aging. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1307–1316.
- Bodega, G.; Alique, M.; Bohórquez, L.; Ciordia, S.; Mena, M.C.; Ramírez, M.R. The Antioxidant Machinery of Young and Senescent Human Umbilical Vein Endothelial Cells and Their Microvesicles. Oxid. Med. Cell Longev. 2017, 2017, 7094781.
- Bodega, G.; Alique, M.; Bohórquez, L.; Morán, M.; Magro, L.; Puebla, L.; Ciordia, S.; Mena, M.C.; Arza, E.; Ramirez, M.R. Young and Especially Senescent Endothelial Microvesicles Produce NADPH: The Fuel for Their Antioxidant Machinery. Oxid. Med. Cell. Longev. 2018, 2018, 12.
- Yáñez-Mó, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell Vesicles 2015, 4, 27066.
- Bodega, G.; Alique, M.; Puebla, L.; Carracedo, J.; Ramírez, R.M. Microvesicles: ROS scavengers and ROS producers. J. Extracell. Vesicles 2019, 8, 1626654.
- Van der Vorst, E.P.C.; de Jong, R.J.; Donners, M.M.P.C. Message in a Microbottle: Modulation of Vascular Inflammation and Atherosclerosis by Extracellular Vesicles. Front. Cardiovasc. Med. 2018, 5, 2.
- Boulanger, C.M.; Loyer, X.; Rautou, P.E.; Amabile, N. Extracellular vesicles in coronary artery disease. Nat. Rev. Cardiol. 2017, 14, 259–272.
- Nomura, S. Extracellular vesicles and blood diseases. Int. J. Hematol. 2017, 105, 392–405.
- New, S.E.; Aikawa, E. Role of extracellular vesicles in de novo mineralization: An additional novel mechanism of cardiovascular calcification. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1753–1758.