Insulin-like growth factor 1 receptor (IGF1R) is a receptor tyrosine kinase that regulates cell growth and proliferation. Upregulation of the IGF1R pathway constitutes a common paradigm shared with other receptor tyrosine kinases such as EGFR, HER2, and MET in different cancer types, including colon cancer. The main IGF1R signaling pathways are PI3K-AKT and MAPK-MEK. However, different processes, such as post-translational modification (SUMOylation), epithelial-to-mesenchymal transition (EMT), and microenvironment complexity, can also contribute to intrinsic and acquired resistance.
- insulin-like growth factor receptor
- metastatic colorectal cancer
1. Insulin and the IGF Pathway
Insulin and insulin-like growth factor 1 (IGF1) are closely related ligands that interact with the insulin receptor (INSR) and IGF1 receptor (IGF1R) family of receptors. Insulin is considered to be a metabolic hormone activating INSR, whereas IGF1 is generally considered to be a growth factor activating IGF1R. IGF1R is a homodimeric, type II receptor tyrosine kinase comprising two extracellular α subunits and two β subunits. The extracellular α subunits are required for IGF1, IGF2, and insulin ligand binding, whereas the two β subunits contain an extracellular portion, transmembrane region, and intracellular tyrosine kinase catalytic domain and ATP-binding site. IGF1R is closely related to INSR, with sequence similarity varying from 41% to 84%, depending on the domain. The interaction of the ligands with IGF1R, INSR, or hybrid receptors activates numerous downstream pathways within the cell. Ligand-activated IGF1R first binds to intracellular adaptor proteins, predominantly insulin receptor substrates (IRS1–6) and Src homology 2 domain-containing transforming protein 1 (SHC). Subsequent phosphorylation of these proteins induces the activation of phosphoinositide 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) pathways [1][2][3]. In addition, IGF1R signals different nuclear transcription factors that contribute, through signal transducers and activators of transcription (STAT) proteins [4], to epithelial-to-mesenchymal transition (EMT), cell survival via upregulation of anti-apoptotic Bcl-XL [5], and adaptive metabolic reprogramming via promotion of glycolysis and glutamine consumption through MYC activation [6].
The bioavailability of IGF ligands is abnormally high in many cancers, and IGF-binding proteins (IGFBPs) and IGFBP proteases are important for regulating ligand bioavailability [7]. Whereas IGF1R mutations have not been described in tumors, IGF1R mutations have been described as rare causes of intrauterine and postnatal growth disorders [8][9]. Polymorphisms in genes encoding either IGF1 or IGFBPs contribute to up- or downregulation of IGF1R function [10]. Upregulation of IGF1R and/or INSR constitutes a common paradigm mediating tumor resistance [11][12] in different cancer types and is associated with poor prognosis in prostate cancer, squamous cell lung cancer, and prostate cancer [13][14][15].
2. Mechanism of Resistance Related to the IGF1R Pathway
2.1. IGF1R Resistance Due to Other Tyrosine Kinase Receptors and Metalloproteinases
IGF1R interacts with tumor suppressor genes and proto-oncogenes. Disturbances in components in the p53/MDM2 network may upregulate IGF1R and confer cancer cells with a growth advantage [16][17][18]. Compensatory signaling mediated by IGF2 through the INRS can contribute to resistance to IGF1R compounds [19]. IGFBP proteins are tightly regulated by serine proteases and matrix metalloproteinases, particularly matrix metalloproteinase-7 (MMP7) [20][21][22][23], which is associated with poor prognosis in metastatic colorectal cancer (mCRC) [24][25]. MMP7 can indirectly contribute to IGFBP degradation [26] and IGF1R-related chemoresistance through AKT-dependent [27] and -independent pathways [28].
2.2. IGF1R, SUMOylation, and Other Post-Translational Procesess
There is functional crosstalk between the conjugation of SUMO to a substrate and other post-translational modifiers, mainly, ubiquitylation, acetylation, and phosphorylation. The interaction between ubiquitylation and SUMOylation is complex and SUMO and ubiquitin are often conjugated to the same substrate, lysine, acting antagonistically or sequentially [36]. Another post-translational modification that was recently identified as a SUMO regulator is acetylation. Acetylation of Ubc9 at residue K65 regulates the SUMOylation of some substrates [37]. There is also a complex interaction between SUMOylation and phosphorylation [38]. Phosphorylation of SUMO substrates can positively or negatively regulate their modification by SUMO, depending on the protein. On the other hand, SUMO can also regulate the phosphorylation system through the SUMOylation of kinases and phosphatases. SUMO proteins control cellular signaling networks and regulate, among others, DNA repair, differentiation, apoptosis, nuclear transport, and EMT differentiation [39][40][41][42][43].
Phosphorylation of IGF1R and its subsequent nuclear translocation has been reported to be mediated by SUMOylation [44]. IGF1R exhibits a different function when phosphorylated in the nucleus because it acts as a transcription regulator (instead of a tyrosine kinase receptor) and its nuclear location is associated with poor prognosis [45]. RANBP2 and PIAS3, the main SUMO E3 ligases, have been implicated in IGF1R nuclear accumulation [46][47]. In the nucleus, pIGF1R has AKT/MEK-independent properties, such as cyclin D1 and axin2 activation [48][49]. Interestingly, treatment of oxaliplatin-resistant colorectal cancer (CRC) cells, but not naïve cells, with IGF1R inhibitors (either ganitumab, a monoclonal antibody, or AEV-541, a tyrosine kinase inhibitor) promoted IGF1R nuclear internalization and PIAS3 inhibition decreased cytoplasm–nuclear IGF1R trafficking [47].
2.3. IGF1R and EMT
EMT is a well-known mechanism of intrinsic [50][51] and acquired [52] chemoresistance. IGF1R signaling was recently implicated in EMT induction/maintenance through STAT3/NANOG/Slug [53] but also, importantly, was found to contribute to acquired chemoresistance [54][55]. Although IGF1R and EMT markers have been shown to be upregulated in drug-tolerant cells, the exact mechanism of resistance remains unknown. In addition, IGF-alternative mechanisms of EMT induction include NF-KB activation [56], WNT/β-catenin [57], and TGFβ/SMAD activation [58], extensively reviewed by Li et al. [59]. Therefore, because other alternatives that are at least partially IGF1R-independent promote EMT, combinations of EMT inhibitors with IGF1R inhibitors are more likely to be effective in CRC.
2.4. IGF1R and the Microenvironment
The tumor microenvironment includes cancer-associated fibroblasts (CAFs) and vascular and immune cells. CAFs and myeloid cells contribute to STAT3 activation in cancer cells through IL-6, CCL2, TGFB, and CCL5 release, and promote regulatory T-cell (Treg) expansion [60]. Preclinical data concerning the combination of IGF1R and STAT3 inhibitors, but not IGF1R inhibition alone, showed a reduction in tumor burden through CAF and myeloid cell depletion [60][61]. Interestingly, anti-IGF1R therapies (cixutumumab) or radiotherapy can promote an immune-suppressive microenvironment due to IGF2- [61] or CAF-dependent IGF1 release from cancer cells [62]. In this latter work, radiotherapy-activated CAFs promoted the survival of CRC cells through metabolic reprogramming (favoring increased glycolysis and glutamine consumption). Therefore, this seminal paper indicates that paracrine IGF/IGF1R signaling contributes to metabolic reprogramming (induction of lactate and glutamine consumption) [63][64][65], a well-known mechanism in the immune-suppressive tumor microenvironment, due to M2 polarization [66] and Treg selection [67].
3. IGF1R Pathway: Lessons to Learn for Adequate Drug Development in RAS Wild-Type mCRC Patients
3.1. IGF1R in the Clinical Scenario
Table 1.
| Author | Study Design | Treatment Arms | LB/N | Biomarker Methodology | Biomarker | Conclusion |
|---|
| Winder et al. [10] | Retrospective/prospective | Cetuximab | No/130 | Polymorphisms | IGF1 (rs2946834-AA) | Increased efficacy |
| Codony-Servat et al. [47] | Retrospective/prospective | CHT+/−mAb | No/470 | IHC | High pIGF1R (nuclear) | Decreased efficacy |
| Fuchs et al. [69] | Retrospective/prospective | CHT | Yes/527 | ELISA | High IGFBP3 and IGF1 | Improved OS |
| García-Albéniz et al. [70] | Retrospective | CHT | Yes/41 | ELISA | IGF1 increment | Improved OS |
| Van Cutsem et al. [71] | Retrospective/prospective | Ganitumab-Panitumumab vs Panitumumab | Yes/94 | ELISA | High IGFBP1 and IGFBP2 | Improved OS |
| Guercio et al. [72] | Prospective | CHT+mAb | Yes/1084 | ELISA | High IGFBP3 and IGFBP7 | Improved OS |
| Scartozzi et al. [73] | Retrospective | I-Cetuximab | No/112 | IHC | Low IGF1 | Increased efficacy |
| Sclafani et al. [74] | Retrospective/prospective | Dalotozumab-Cetuximab-I vs Cetuximab-I | No/344 | IHC | High IGF1 | Decreased OS |
| Huang et al. [75] | Retrospective | Cetuximab | No/70 | RNA expression | High IGF1R | Increased efficacy |
CHT, chemotherapy; LB, liquid biopsy; N, number of patients included; I, irinotecan; IHC, immunohistochemistry; mAb, monoclonal antibody; pIGF1R, phosphorylated IGF1R.
Our group and others have evaluated potential IGF biomarkers based on liquid biopsy (ligands (IGF or IGFBP) by ELISA) [69][70][71][72] or IGF, IGFBP, and/or IGF1R protein in paraffin-embedded tissues (immunohistochemistry [73][74]), DNA (polymorphisms [10]), or RNA expression [75]. Global interpretation is hampered by trial design (most results are derived from retrospective analysis that were not designed for this purpose), nonstandardized biomarker cut-offs, and the use of different techniques (ELISA, DNA polymorphisms, and RNA expression). However, we conclude that high levels of IGFBP could identify a subgroup of mCRC patients with better prognosis [72]. It remains unclear whether any of these markers would predict the efficacy of IGF1R or EGFR inhibitors.
3.2. Liquid Biopsy in RAS Wild-Type Anti-EGFR Pretreated Patients to Select Optimal Patients for New Therapies. Mechanism of Resistance
The identification of biomarkers of the response to anti-EGFR therapy has proven to be promising. The monoclonal antibodies cetuximab and panitumumab, which neutralize the extracellular domain of EGFR, have demonstrated effectiveness, in terms of both a better response and improved survival when used as first-, second-, or third-line treatment regimens. Unfortunately, however, patients with mCRC who have mutant RAS (KRAS/NRAS; ~55%) or BRAF (V600E; ~5–10%) genes do not derive any therapeutic benefit from anti-EGFR drugs [76][77]. Although patients with wild-type (WT) RAS and BRAF (30–35% of mCRC patients) are sensitive to first-line chemotherapy plus anti-EGFR treatment (best overall response (BOR) range, 55–75%) [78][79][80], most of these patients develop resistance within the first 2 years. Primary inter- and intratumor heterogeneity and acquisition of secondary RAS and BRAF mutations during the course of such treatments are the major factors responsible for acquired resistance [81][82][83][84][85][86][87][88] (
Figure 1). The recent gene expression-based molecular classification of consensus molecular subtypes (CMSs) has demonstrated the existence of phenotypic heterogeneity in CRC [89], highlighting the need for improved risk stratification and selection of patient populations before any specific treatment options are offered. Although the CMS classification in mCRC suggested prognostic significance, its clinical significance in terms of predicting the response to anti-EGFR therapy remains unclear [90][91][92]. Furthermore, given that genetic mutations can induce certain gene expression phenotypes in CRC, a comprehensive phenotypic exploration (including not only cancer epithelial cells, but also the tumor cell-type microenvironment) is critically required to establish biomarkers that can robustly predict the response to anti-EGFR therapies.
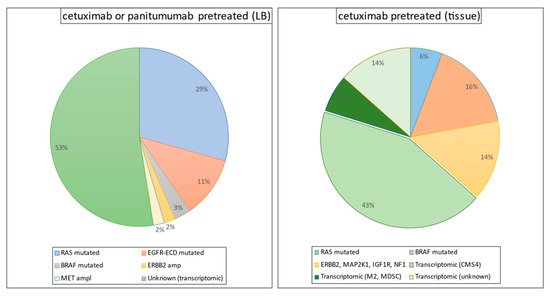
Figure 1.
3.3. New Targeted Agents in Anti-EGFR Pretreated Patients with RAS Wild-Type mCRC
3.3.1. Selection of Optimal Patients with Liquid Biopsy (RAS and BRAF)
CRC patients included in clinical trials of anti-IGF1R compounds and Sym004 (a mixture of two monoclonal antibodies—futuximab and modotuximab—that binds epitopes in the EGFR extracellular domain) were not selected based on RAS/BRAF WT in liquid biopsy [8][71][93][94]. This aspect is important because 30–35% of included patients basally had RAS or BRAF mutations that are related to intrinsic EGFR resistance [94]. Therefore, we propose that new agents or re-challenge strategies in patients progressing to anti-EGFR include only patients with liquid biopsy determination of the RAS and BRAF genotype. In ongoing EGFR re-challenge studies or those with new compounds (anti-HER2 and anti-MET), liquid biopsy is not mandatory to exclude RAS/BRAF patients. We believe that this limitation can compromise the efficacy of new drugs.
3.3.2. Prospective Trials with New Strategies or New Compounds (HER2 Inhibitors, MET Inhibitors, and Re-Challenge with EGFR Inhibitors [Cetuximab or Panitumumab])
The IGF axis has been proposed as a target for anticancer therapies. Antibody, tyrosine kinase, and ligand inhibitors of the IGF receptor have been studied. In phase I trials, these antibodies seem to be well-tolerated; the most common toxicity is hyperglycemia [95]. Early-phase trials in the last lines of treatment showed promising results, mainly a stable response, but the results were negative in later-phase trials. The efficacy of anti-IGF1R and Sym004 was hugely disappointing, with nearly null activity with anti-IGF1R compounds (ganitumab, figitumumab, and dalotuzumab) and less than a 15% response rate with Sym004 [71][74][93][94]. One of the major limitations in these studies is the absence of targeted biomarker selection. This is a potentially important issue to address with new strategies (re-challenge with EGFR compounds [96]) or new agents (HER2 and MET inhibitors). Re-challenge strategies lack a suitable biomarker for patient selection, but only patients with HER2+++ overexpression (with DS8201) or patients with MET amplification should probably be included with new agents. The importance of biomarker selection has been highlighted by the low activity of MET inhibitors without biomarker selection [97][98].
RAS
BRAF mutation (2WT) mCRC patients with HER2 overexpression or amplification [99][100][101][102][103] (
Table 2.
| Author | Design(n) | Treatment arms | LLB at Entry * | Biomarker Driven **/Targeted Therapy BiBiomarker Driven **/Targeted Therapyomarker Selection | BOR% | PFS (m) | mOS (m) | ESMO-MCBS: ESMO-Magnitude Clinical Benefit Scale |
|---|
| Montagut et al. [94] | II-R (254) | Sym004-12 mg/kg vs. Sym004-9 mg/kg vs. Investigator choice | No | No | 14.1 9.6 2.9 |
- - - |
11.9 8.9 8.4 |
1 |
| Cremolini et al. [96] | II (28) | Irinotecan-Cetuximab | No | No | 14 | 4.1 ** | - | NA |
| Rimassa et al. [97] | II (41) | Tivantinib-Cetuximab | No | No | 9.8 | 2.6 | - | NA |
| Delord et al. [98] | I-II (13) | Capmatinib-Cetuximab | No | No | 0 | - | - | NA |
| Sartore-Bianchi et al. [99] | II (27) | Trastuzumab-Lapatinib | No | Yes/HER2 (+++) | 26 | 5.1 | - | NA |
| Nakamura et al. [100] | II (18) | Trastuzumab-Pertuzumab | Yes | Yes/HER2 (amplification) | 35 | 4 | - | NA |
| Gupta et al. [101] | II (28) | Trastuzumab-Pertuzumab | No | Yes/HER2 (+++) | 25 | 4.2 | - | NA |
| Sartore-Bianchi et al. [102] | II (31) | TDM1-Pertuzumab | No | Yes/HER2 (+++) | 9.7 | 4.1 | - | NA |
| Siena et al. [103] | II (53) | Trastuzumab-Deruxtecan | No | Yes/HER2 (+++) | 43.4 | 6.9 | - | NA |
| CHRONOS * | II (129) | Panitumumab | Yes | No | - | - | - | - |
| BEYOND * | II-R (85) | FOLFIRI-Panitumumab vs FOLFIRI | Yes | No | - | - | - | - |
| VELO * | II-R (112) | TAS-102-Panitumumab vs TAS-102 | No | No | - | - | - | - |
| PULSE * | II-R (106) | Panitumumab vs Regorafenib or TAS-102 | Yes | No | - | - | - | - |
| FIRE-4 * | III (230) | Irinotecan-Cetuximab vs Regorafenib or Investigator choice | No | No | - | - | - | - |
| PERSPECTIVE * | II (48) | Tepotinib-Cetuximab | Yes | Yes/MET (amplification) | - | - | - | - |
2WT, double wild-type for RAS and BRAF mutations; N, number; LB, liquid biopsy; BOR, best overall response; mPFS, median progression-free survival in months; mOS, median overall survival in months; ESMO-MCBS, ESMO-Magnitude Clinical Benefit Scale; NA, not applicable. * Prospective studies without available results. ** Patients without mutations in liquid biopsy. (+++) Amplification 3 crosses.
MET inhibitors (capmatinib and tivantinib) have been tested in combination with cetuximab in two phase II studies [97][98]. Globally, in nonselected patients, the activity is very modest with a BOR < 10% and a less than 3-month median PFS. An unplanned subanalysis with tivantinib suggested that MET-amplified patients would be more sensitive to MET inhibition. Due to this subanalysis, a prospective clinical trial has recently begun recruiting mCRC patients with MET amplification (PERSPECTIVE). Although this trial will include only mCRC patients with MET amplification, other well-known resistance mechanisms such as EMT (which increases not only hepatocyte growth factor, but also other alternative EMT-driven biomarkers) would also contribute to tepotinib-cetuximab resistance [105].
3.3.3. EGFR and Trastuzumab-Acquired Resistance
In addition to genetic mutations, a comprehensive profiling of gene expression patterns and their application in tissue and plasma samples might not only allow identification of superior predictive biomarkers for anti-EGFR therapy, but also further understanding of potential new strategies (such as re-challenge with anti-EGFR) or the addition of new agents. Indeed, less than 30% of patients that progress to chemotherapy plus anti-EGFR compounds can currently be explained by genetic resistance mechanisms and detected by ctDNA analysis. A transcriptomic resistance mechanism to cetuximab has recently been described, emphasizing the importance of the immune microenvironment in anti-EGFR resistance [88]. Cetuximab and panitumumab exert at least part of their activity through Fc-mediated antibody-dependent cellular cytotoxicity (ADCC), although this mechanism of resistance has largely been unexplored. ADCC activity is hampered by M2 microenvironment polarization [106][107] or antibody-dependent cellular phagocytosis (ADCP) loss mediated by dendritic cells [108] and, therefore, deciphering immune microenvironment changes could potentially improve current anti-EGFR efficacy.
Trastuzumab activity in preclinical models has recently been associated with ADCP mediated by macrophages but not with ADCC through natural killer cells or neutrophils, complement cellular cytotoxicity (CDC), or T-cell adaptive immunity [109]. CD47 overexpression in cancer cells (a “do not eat me” receptor) has been inversely associated with trastuzumab efficacy. DS-8201a, also in a preclinical CRC mouse model, upregulates CD86, dendritic cells, and CD8 T-cells and PD-L1 and MHC-I in tumor cells, which emphasizes the importance of not only ADCP, but also adaptive immunity in drug activity. It seems that its crucial effect on immune activation is specifically due to the effect of deruxtecan [110]. Despite these preclinical data with trastuzumab and DS8201a, only 45% of patients clinically responded to DS8201a and less than 30% to trastuzumab plus lapatinib or pertuzumab. In addition, most initial responders (>90% of cases) progress in the first year. Therefore, understanding of how metabolic reprogramming can impair ADCP function or how chronic therapeutic antibody-mediated ADCP stimulation can influence acquired mechanisms of trastuzumab or DS8201a resistance would be valuable. Autophagy, a metabolic reprogramming process increased in highly hypoxic nutrient-deprived tumors such as pancreatic adenocarcinomas [111], has recently been shown to increase immune suppression [112][113]. It is further unclear how autophagy suppresses the immune system, although Yamamoto et al. suggest that autophagy increases immunosuppression through reduced MCH-I expression [112]. Other authors indicate that autophagy can decrease TNFα-dependent cell death by immune CD8+ T-cells [113]. We are tempted to speculate that continuous ADCP stimulation (either by cancer-mediated autophagy metabolic reprogramming or antibody-mediated stimuli) induces LAP-dependent M2 polarization [114] and PD-L1 and IDO-1 expression [115] (see
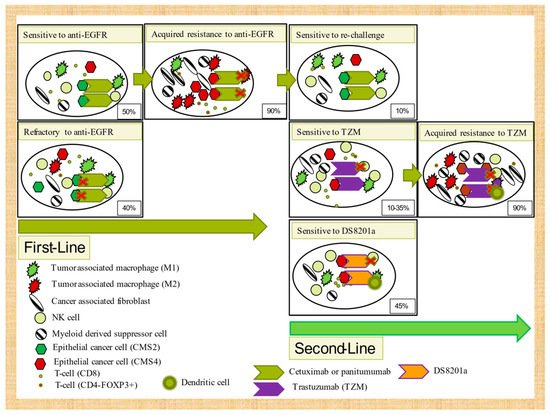
Figure 2.