Diffuse large B-cell lymphomas (DLBCL)s, the most common type of Non-Hodgkin’s Lymphoma, constitute a heterogeneous group of disorders including different disease sites, strikingly diverse molecular features and a profound variability in the clinical behavior. Molecular studies and clinical trials have partially revealed the underlying causes for this variability and have made possible the recognition of some molecular variants susceptible of specific therapeutic approaches. The main histogenetic groups include the germinal center, activated B cells, thymic B cells and terminally differentiated B cells, a basic scheme where the large majority of DLBCL cases can be ascribed. The nodal/extranodal origin, specific mutational changes and microenvironment peculiarities provide additional layers of complexity.
- large B-cell lymphoma
- targeted therapy
- molecular classification
1. Introduction
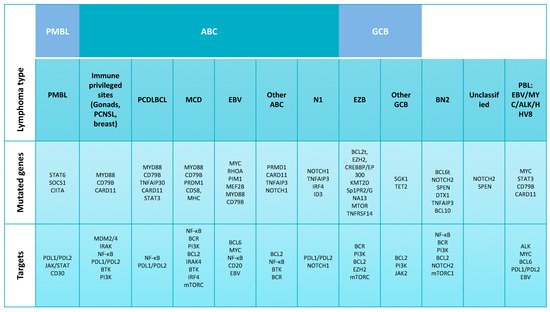
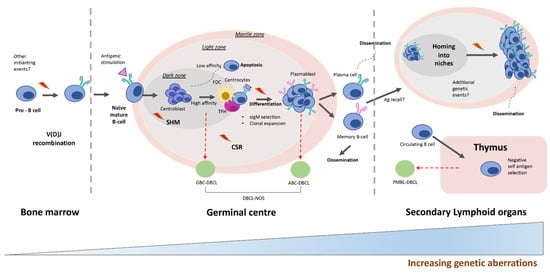
2. Molecular Alterations Defining Aggressive DLBCL
2.1. DLBCL Subclassification
2.2. Relevant Genes and Pathways
3. B-Cell Receptor Signaling and Toll-Like Receptor Pathways
3.1. BCR
3.2. Toll-Like Receptor Signaling
3.3. NF-κB Pathway
3.4. PI3K/AKT/mTOR Pathway
4. Therapeutic Targeting of Diffuse Large B-Cell Lymphoma
Molecular analysis and clinical trials in DLBCL are progressively revealing multiple therapeutic opportunities that eventually could replace the currently used chemotherapy. These new options are tumor-type specific and, thus, adapted for the GC, ABC, thymic or terminally differentiated phenotypes and, additionally, in many cases, are coupled with precise genetic events or deregulated pathways.4.1. BCL6 Inhibitors
4.2. BCL2/MYC Inhibitors
4.3. BTK iInhibition
4.4. Toll-Like Receptor Inhibition
4.5. PI3K Inhibition
4.6. NF-κB Inhibition
4.7. JAK/STAT Inhibition
4.8. ICIs
References
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390.
- Sujobert, P.; Salles, G.; Bachy, E. Molecular Classification of Diffuse Large B-cell Lymphoma: What Is Clinically Relevant? Hematol. Oncol. Clin. N. Am. 2016, 30, 1163–1177.
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511.
- Wright, G.; Tan, B.; Rosenwald, A.; Hurt, E.H.; Wiestner, A.; Staudt, L.M. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 2003, 100, 9991–9996.
- Lenz, G.; Wright, G.W.; Emre, N.C.T.; Kohlhammer, H.; Dave, S.S.; Davis, R.E.; Carty, S.; Lam, L.T.; Shaffer, A.L.; Xiao, W.; et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 13520–13525.
- Rosenwald, A.; Wright, G.; Chan, W.C.; Connors, J.M.; Campo, E.; Fisher, R.I.; Gascoyne, R.D.; Muller-Hermelink, H.K.; Smeland, E.B.; Giltnane, J.M.; et al. The Use of Molecular Profiling to Predict Survival after Chemotherapy for Diffuse Large-B-Cell Lymphoma. N. Engl. J. Med. 2002, 346, 1937–1947.
- Xu, J.; Liu, J.L.; Medeiros, L.J.; Huang, W.; Khoury, J.D.; McDonnell, T.J.; Tang, G.; Schlette, E.; Yin, C.C.; Bueso-Ramos, C.E.; et al. MYC rearrangement and MYC/BCL2 double expression but not cell-of-origin predict prognosis in R-CHOP treated diffuse large B-cell lymphoma. Eur. J. Haematol. 2020, 104, 336–343.
- Abdulla, M.; Hollander, P.; Pandzic, T.; Mansouri, L.; Ednersson, S.B.; Andersson, P.; Hultdin, M.; Fors, M.; Erlanson, M.; Degerman, S.; et al. Cell-of-origin determined by both gene expression profiling and immunohistochemistry is the strongest predictor of survival in patients with diffuse large B-cell lymphoma. Am. J. Hematol. 2020, 95, 57–67.
- Scott, D.W.; Mottok, A.; Ennishi, D.; Wright, G.W.; Farinha, P.; Ben-Neriah, S.; Kridel, R.; Barry, G.S.; Hother, C.; Abrisqueta, P.; et al. Prognostic Significance of Diffuse Large B-Cell Lymphoma Cell of Origin Determined by Digital Gene Expression in Formalin-Fixed Paraffin-Embedded Tissue Biopsies. J. Clin. Oncol. 2015, 33, 2848–2856.
- Nowakowski, G.S.; Feldman, T.; Rimsza, L.M.; Westin, J.R.; Witzig, T.E.; Zinzani, P.L. Integrating precision medicine through evaluation of cell of origin in treatment planning for diffuse large B-cell lymphoma. Blood Cancer J. 2019, 9, 48.
- Staiger, A.M.; Ziepert, M.; Horn, H.; Scott, D.W.; Barth, T.F.; Bernd, H.W.; Feller, A.C.; Klapper, W.; Szczepanowski, M.; Hummel, M.; et al. Clinical Impact of the Cell-of-Origin Classification and the MYC/BCL2 Dual Expresser Status in Diffuse Large B-Cell Lymphoma Treated Within Prospective Clinical Trials of the German High-Grade Non-Hodgkin’s Lymphoma Study Group. J. Clin. Oncol. 2017, 35, 2515–2526.
- Younes, A.; Sehn, L.H.; Johnson, P.; Zinzani, P.L.; Hong, X.; Zhu, J.; Patti, C.; Belada, D.; Samoilova, O.; Suh, C.; et al. Randomized Phase III Trial of Ibrutinib and Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone in Non–Germinal Center B-Cell Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2019, 37, 1285–1295.
- Nowakowski, G.S.; Chiappella, A.; Witzig, T.E.; Spina, M.; Gascoyne, R.D.; Zhang, L.; Flament, J.; Repici, J.; Vitolo, U. ROBUST: Lenalidomide-R-CHOP versus placebo-R-CHOP in previously untreated ABC-type diffuse large B-cell lymphoma. Future Oncol. 2016, 12, 1553–1563.
- Davies, A.; Cummin, T.E.; Barrans, S.; Maishman, T.; Mamot, C.; Novak, U.; Caddy, J.; Stanton, L.; Kazmi-Stokes, S.; McMillan, A.; et al. Gene-expression profiling of bortezomib added to standard chemoimmunotherapy for diffuse large B-cell lymphoma (REMoDL-B): An open-label, randomised, phase 3 trial. Lancet Oncol 2019, 20, 649–662.
- Sáez, A.I.; Sáez, A.J.; Artiga, M.J.; Pérez-Rosado, A.; Camacho, F.I.; Díez, A.; García, J.F.; Fraga, M.; Bosch, R.; Rodríguez-Pinilla, S.M.; et al. Building an outcome predictor model for diffuse large B-cell lymphoma. Am. J. Pathol. 2004, 164, 63150–63151.
- Sanchez, E.; Chacon, I.; Plaza, M.M.; Muñoz, E.; Cruz, M.A.; Martinez, B.; Lopez, L.; Martinez-Montero, J.C.; Orradre, J.L.; Saez, A.I.; et al. Clinical outcome in diffuse large B-cell lymphoma is dependent on the relationship between different cell-cycle regulator proteins. J. Clin. Oncol. 1998, 16, 1931–1939.
- Batlle-López, A.; de Villambrosía, S.G.; Francisco, M.; Malatxeberria, S.; Sáez, A.; Montalban, C.; Sánchez, L.; Garcia, J.F.; González-Barca, E.; López-Hernández, A.; et al. Stratifying diffuse large B-cell lymphoma patients treated with chemoimmunotherapy: GCB/non-GCB by immunohistochemistry is still a robust and feasible marker. Oncotarget 2016, 7, 18036–18049.
- Hu, S.; Xu-Monette, Z.Y.; Tzankov, A.; Green, T.; Wu, L.; Balasubramanyam, A.; Liu, W.-M.; Visco, C.; Li, Y.; Miranda, R.N.; et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: A report from The International DLBCL Rituximab-CHOP Consortium Program. Blood 2013, 121, 4021–4031.
- Rosenwald, A.; Bens, S.; Advani, R.; Barrans, S.; Copie-Bergman, C.; Elsensohn, M.-H.; Natkunam, Y.; Calaminici, M.; Sander, B.; Baia, M.; et al. Prognostic Significance of MYC Rearrangement and Translocation Partner in Diffuse Large B-Cell Lymphoma: A Study by the Lunenburg Lymphoma Biomarker Consortium. J. Clin. Oncol. 2019, 37, 3359–3368.
- Scott, D.W.; King, R.L.; Staiger, A.M.; Ben-Neriah, S.; Jiang, A.; Horn, H.; Mottok, A.; Farinha, P.; Slack, G.W.; Ennishi, D.; et al. High-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements with diffuse large B-cell lymphoma morphology. Blood 2018, 131, 2060–2064.
- Johnson, N.A.; Slack, G.W.; Savage, K.J.; Connors, J.M.; Ben-Neriah, S.; Rogic, S.; Scott, D.W.; Tan, K.L.; Steidl, C.; Sehn, L.H.; et al. Concurrent Expression of MYC and BCL2 in Diffuse Large B-Cell Lymphoma Treated with Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone. J. Clin. Oncol. 2012, 30, 3452–3459.
- Beltran, B.E.; Castro, D.; Paredes, S.; Miranda, R.N.; Castillo, J.J. EBV-positive diffuse large B-cell lymphoma, not otherwise specified: 2020 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2020, 95, 435–445.
- Xu-Monette, Z.Y.; Tu, M.; Jabbar, K.J.; Cao, X.; Tzankov, A.; Visco, C.; Cai, Q.; Montes-Moreno, S.; An, Y.; Dybkaer, K.; et al. Clinical and biological significance of de novo CD5+ diffuse large B-cell lymphoma in Western countries. Oncotarget 2015, 6, 5615–5633.
- Ok, C.Y.; Li, L.; Xu-Monette, Z.Y.; Visco, C.; Tzankov, A.; Manyam, G.C.; Montes-Moreno, S.; Dybaer, K.; Chiu, A.; Orazi, A.; et al. Prevalence and Clinical Implications of Epstein-Barr Virus Infection in De Novo Diffuse Large B-Cell Lymphoma in Western Countries. Clin. Cancer Res. 2014, 20, 2338–2349.
- Montes-Moreno, S.; Odqvist, L.; Diaz-Perez, J.A.; Lopez, A.B.; de Villambrosía, S.G.; Mazorra, F.; Castillo, M.E.; Lopez, M.; Pajares, R.; García, J.F.; et al. EBV-positive diffuse large B-cell lymphoma of the elderly is an aggressive post-germinal center B-cell neoplasm characterized by prominent nuclear factor-kB activation. Mod. Pathol. 2012, 25, 968–982.
- Menter, T.; Bodmer-Haecki, A.; Dirnhofer, S.; Tzankov, A. Evaluation of the diagnostic and prognostic value of PDL1 expression in Hodgkin and B-cell lymphomas. Hum. Pathol. 2016, 54, 17–24.
- Gravelle, P.; Burroni, B.; Péricart, S.; Rossi, C.; Bezombes, C.; Tosolini, M.; Damotte, D.; Brousset, P.; Fournié, J.-J.; Laurent, C. Mechanisms of PD-1/PD-L1 expression and prognostic relevance in non-Hodgkin lymphoma: A summary of immunohistochemical studies. Oncotarget 2017, 8, 44960–44975.
- Hu, S.; Xu-Monette, Z.Y.; Balasubramanyam, A.; Manyam, G.C.; Visco, C.; Tzankov, A.; Liu, W.-M.; Miranda, R.N.; Zhang, L.; Montes-Moreno, S.; et al. CD30 expression defines a novel subgroup of diffuse large B-cell lymphoma with favorable prognosis and distinct gene expression signature: A report from the International DLBCL Rituximab-CHOP Consortium Program Study. Blood 2013, 121, 2715–2724.
- Mottok, A.; Wright, G.; Rosenwald, A.; Ott, G.; Ramsower, C.; Campo, E.; Braziel, R.M.; Delabie, J.; Weisenburger, D.D.; Song, J.Y.; et al. Molecular classification of primary mediastinal large B-cell lymphoma using routinely available tissue specimens. Blood 2018, 132, 2401–2405.
- Campo, E.; Swerdlow, S.H.; Harris, N.L.; Pileri, S.; Stein, H.; Jaffe, E.S. The 2008 WHO classification of lymphoid neoplasms and beyond: Evolving concepts and practical applications. Blood 2011, 117, 5019–5032.
- Mitteldorf, C.; Berisha, A.; Pfaltz, M.C.; Broekaert, S.M.; Schön, M.P.; Kerl, K.; Kempf, W. Tumor Microenvironment and Checkpoint Molecules in Primary Cutaneous Diffuse Large B-Cell Lymphoma—New Therapeutic Targets. Am. J. Surg. Pathol. 2017, 41, 998–1004.
- Mareschal, S.; Pham-Ledard, A.; Viailly, P.J.; Dubois, S.; Bertrand, P.; Maingonnat, C.; Fontanilles, M.; Bohers, E.; Ruminy, P.; Tournier, I.; et al. Identification of Somatic Mutations in Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type by Massive Parallel Sequencing. J. Investig. Dermatol. 2017, 137, 1984–1994.
- Maciocia, P.; Badat, M.; Cheesman, S.; D’Sa, S.; Joshi, R.; Lambert, J.; Mohamedbhai, S.; Pule, M.; Linch, D.; Ardeshna, K. Treatment of diffuse large B-cell lymphoma with secondary central nervous system involvement: Encouraging efficacy using CNS-penetrating R-IDARAM chemotherapy. Br. J. Haematol. 2015, 172, 545–553.
- Monti, S. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood 2005, 105, 1851–1861.
- Gebauer, N.; Gebauer, J.; Hardel, T.T.; Bernard, V.; Biersack, H.; Lehnert, H.; Rades, D.; Feller, A.C.; Thorns, C. Prevalence of targetable oncogenic mutations and genomic alterations in Epstein–Barr virus-associated diffuse large B-cell lymphoma of the elderly. Leuk. Lymphoma 2015, 56, 1100–1106.
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690.
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407.
- Lacy, S.E.; Barrans, S.L.; Beer, P.A.; Painter, D.; Smith, A.G.; Roman, E.; Cooke, S.L.; Ruiz, C.; Glover, P.; van Hoppe, S.J.L.; et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: A Haematological Malignancy Research Network report. Blood 2020, 135, 1759–1771.
- Gonzalez-Aguilar, A.; Idbaih, A.; Boisselier, B.; Habbita, N.; Rossetto, M.; Laurenge, A.; Bruno, A.; Jouvet, A.; Polivka, M.; Adam, C.; et al. Recurrent Mutations of MYD88 and TBL1XR1 in Primary Central Nervous System Lymphomas. Clin. Cancer Res. 2012, 18, 5203–5211.
- Ollila, T.A.; Olszewski, A.J. Extranodal Diffuse Large B Cell Lymphoma: Molecular Features, Prognosis, and Risk of Central Nervous System Recurrence. Curr. Treat. Options Oncol. 2018, 19, 38.
- Hoefnagel, J.J.; Dijkman, R.; Basso, K.; Jansen, P.M.; Hallermann, C.; Willemze, R.; Tensen, C.P.; Vermeer, M.H. Distinct types of primary cutaneous large B-cell lymphoma identified by gene expression profiling. Blood 2005, 105, 3671–3678.
- Pham-Ledard, A.; Cappellen, D.; Martinez, F.; Vergier, B.; Beylot-Barry, M.; Merlio, J.-P. MYD88 Somatic Mutation Is a Genetic Feature of Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type. J. Investig. Dermatol. 2012, 132, 2118–2120.
- Ambrosio, M.R.; Piccaluga, P.P.; Ponzoni, M.; Rocca, B.J.; Malagnino, V.; Onorati, M.; de Falco, G.; Calbi, V.; Ogwang, M.; Naresh, K.N.; et al. The Alteration of Lipid Metabolism in Burkitt Lymphoma Identifies a Novel Marker: Adipophilin. PLoS ONE 2012, 7, e44315.
- Pasqualucci, L. Molecular pathogenesis of germinal center-derived B cell lymphomas. Immunol. Rev. 2019, 288, 240–261.
- Krull, J.E.; Wenzl, K.; Hartert, K.T.; Manske, M.K.; Sarangi, V.; Maurer, M.J.; Larson, M.C.; Nowakowski, G.S.; Ansell, S.M.; McPhail, E.; et al. Somatic copy number gains in MYC, BCL2, and BCL6 identifies a subset of aggressive alternative-DH/TH DLBCL patients. Blood Cancer J. 2020, 10, 1–8.
- Küppers, R.; Dalla-Favera, R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene 2001, 20, 5580–5594.
- Pasqualucci, L.; Neumeister, P.; Goossens, T.; Nanjangud, G.; Chaganti, R.S.K.; Küppers, R.; Dalla-Favera, R. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature 2001, 412, 341–346.
- Pangault, C.; Amé-Thomas, P.; Rossille, D.; Dulong, J.; Caron, G.; Nonn, C.; Chatonnet, F.; Desmots, F.; Launay, V.; Lamy, T.; et al. Integrative Analysis of Cell Crosstalk within Follicular Lymphoma Cell Niche: Towards a Definition of the FL Supportive Synapse. Cancers 2020, 12, 2865.
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J.; et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011, 471, 189–195.
- Bakhshi, T.J.; Georgel, P.T. Genetic and epigenetic determinants of diffuse large B-cell lymphoma. Blood Cancer J. 2020, 10, 1–23.
- Reddy, A.; Zhang, J.; Davis, N.S.; Moffitt, A.B.; Love, C.L.; Waldrop, A.; Leppa, S.; Pasanen, A.; Meriranta, L.; Karjalainen-Lindsberg, M.-L.; et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017, 171, 481–494.e15.
- Cader, F.Z.; Schackmann, R.C.J.; Hu, X.; Wienand, K.; Redd, R.; Chapuy, B.; Ouyang, J.; Paul, N.; Gjini, E.; Lipschitz, M.; et al. Mass cytometry of Hodgkin lymphoma reveals a CD4+ regulatory T-cell–rich and exhausted T-effector microenvironment. Blood 2018, 132, 825–836.
- Phelan, J.D.; Young, R.M.; Webster, D.E.; Roulland, S.; Wright, G.W.; Kasbekar, M.; Shaffer, A.L., III; Ceribelli, M.; Wang, J.Q.; Schmitz, R.; et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature 2018, 560, 387–391.
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010, 463, 88–92.
- Pasqualucci, L.; Compagno, M.; Houldsworth, J.; Monti, S.; Grunn, A.; Nandula, S.V.; Aster, J.C.; Murty, V.V.; Shipp, M.A.; Dalla-Favera, R. Inactivation of the PRDM1/BLIMP1 gene in diffuse large B cell lymphoma. J. Exp. Med. 2006, 203, 311–317.
- Shaffer, A.; Lin, K.-I.; Kuo, T.C.; Yu, X.; Hurt, E.M.; Rosenwald, A.; Giltnane, J.M.; Yang, L.; Zhao, H.; Calame, K.; et al. Blimp-1 Orchestrates Plasma Cell Differentiation by Extinguishing the Mature B Cell Gene Expression Program. Immunology 2002, 17, 51–62.
- Lamason, R.L.; McCully, R.R.; Lew, S.M.; Pomerantz, J.L. Oncogenic CARD11 Mutations Induce Hyperactive Signaling by Disrupting Autoinhibition by the PKC-Responsive Inhibitory Domain. Biochemistry 2010, 49, 8240–8250.
- Tibiletti, M.G.; Martin, V.; Bernasconi, B.; del Curto, B.; Pecciarini, L.; Uccella, S.; Pruneri, G.; Ponzoni, M.; Mazzucchelli, L.; Martinelli, G.; et al. BCL2, BCL6, MYC, MALT 1, and BCL10 rearrangements in nodal diffuse large B-cell lymphomas: A multicenter evaluation of a new set of fluorescent in situ hybridization probes and correlation with clinical outcome. Hum. Pathol. 2009, 40, 645–652.
- Knies, N.; Alankus, B.; Weilemann, A.; Tzankov, A.; Brunner, K.; Ruff, T.; Kremer, M.; Keller, U.B.; Lenz, G.; Ruland, J. Lymphomagenic CARD11/BCL10/MALT1 signaling drives malignant B-cell proliferation via cooperative NF-κB and JNK activation. Proc. Natl. Acad. Sci. USA 2015, 112, E7230–E7238.
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.-H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119.
- Iwasaki, A.; Medzhitov, R. Regulation of Adaptive Immunity by the Innate Immune System. Science 2010, 327, 291–295.
- Rovira, J.; Karube, K.; Valera, A.; Colomer, D.; Enjuanes, A.; Colomo, L.; Martínez-Trillos, A.; Giné, E.; Dlouhy, I.; Magnano, L.; et al. MYD88 L265P Mutations, But No Other Variants, Identify a Subpopulation of DLBCL Patients of Activated B-cell Origin, Extranodal Involvement, and Poor Outcome. Clin. Cancer Res. 2016, 22, 2755–2764.
- Davis, R.E.; Brown, K.D.; Siebenlist, U.; Staudt, L.M. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J. Exp. Med. 2001, 194, 1861–1874.
- Lenz, G.; Davis, R.E.; Ngo, V.N.; Lam, L.; George, T.C.; Wright, G.W.; Dave, S.S.; Zhao, H.; Xu, W.; Rosenwald, A.; et al. Oncogenic CARD11 Mutations in Human Diffuse Large B Cell Lymphoma. Science 2008, 319, 1676–1679.
- Boone, D.L.; Turer, E.E.; Lee, E.G.; Ahmad, R.-C.; Wheeler, M.T.; Tsui, C.; Hurley, P.; Chien, M.; Chai, S.; Hitotsumatsu, O.; et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat. Immunol. 2004, 5, 1052–1060.
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 2009, 459, 717–721.
- Wang, J.Q.; Jeelall, Y.S.; Beutler, B.; Horikawa, K.; Goodnow, C.C. Consequences of the recurrent MYD88L265P somatic mutation for B cell tolerance. J. Exp. Med. 2014, 211, 413–426.
- Wenzl, K.; Manske, M.K.; Sarangi, V.; Asmann, Y.W.; Greipp, P.T.; Schoon, H.R.; Braggio, E.; Maurer, M.J.; Feldman, A.L.; Witzig, T.E.; et al. Loss of TNFAIP3 enhances MYD88L265P-driven signaling in non-Hodgkin lymphoma. Blood Cancer J. 2018, 8, 97.
- Young, R.M.; Shaffer, A.L.; Phelan, J.D.; Staudt, L.M. B-Cell Receptor Signaling in Diffuse Large B-Cell lymphoma. Semin. Hematol. 2015, 52, 77–85.
- Uddin, S.; Hussain, A.R.; Siraj, A.K.; Manogaran, P.S.; Al-Jomah, N.A.; Moorji, A.; Atizado, V.; Al-Dayel, F.; Belgaumi, A.; El-Solh, H.; et al. Role of phosphatidylinositol 3′-kinase/AKT pathway in diffuse large B-cell lymphoma survival. Blood 2006, 108, 4178–4186.
- Pfeifer, M.; Grau, M.; Lenze, D.; Wenzel, S.-S.; Wolf, A.; Wollert-Wulf, B.; Dietze, K.; Nogai, H.; Storek, B.; Madle, H.; et al. PTEN loss defines a PI3K/AKT pathway-dependent germinal center subtype of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 12420–12425.
- Kloo, B.; Nagel, D.; Pfeifer, M.; Grau, M.; Düwel, M.; Vincendeau, M.; Dörken, B.; Lenz, P.; Lenz, G.; Krappmann, D. Critical role of PI3K signaling for NF-kappaB-dependent survival in a subset of activated B-cell-like diffuse large B-cell lymphoma cells. Proc. Natl. Acad. Sci. USA 2011, 108, 272–277.
- Su, Y.W.; Hao, Z.; Hirao, A.; Yamamoto, K.; Lin, W.J.; Young, A.; Duncan, G.S.; Yoshida, H.; Wakeham, A.; Lang, P.A.; et al. 14-3-3sigma regulates B-cell homeostasis through stabilization of FOXO1. Proc. Natl. Acad. Sci. USA 2011, 108, 1555–1560.
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nat. Cell Biol. 2011, 476, 298–303.
- Dominguez-Sola, D.; Kung, J.; Holmes, A.B.; Wells, V.A.; Mo, T.; Basso, K.; Dalla-Favera, R. The FOXO1 Transcription Factor Instructs the Germinal Center Dark Zone Program. Immunity 2015, 43, 1064–1074.
- Cerchietti, L.C.; Ghetu, A.F.; Zhu, X.; Da Silva, G.F.; Zhong, S.; Matthews, M.; Bunting, K.L.; Polo, J.M.; Fares, C.; Arrowsmith, C.H.; et al. A small-molecule inhibitor of BCL6 kills DLBCL cells in vitro and in vivo. Cancer Cell 2010, 17, 400–411.
- Cerchietti, L.C.; Lopes, E.C.; Yang, S.N.; Hatzi, K.; Bunting, K.L.; Tsikitas, L.A.; Malik, A.; Robles, A.I.; Walling, J.; Varticovski, L.; et al. A purine scaffold Hsp90 inhibitor destabilizes BCL-6 and has specific antitumor activity in BCL-6-dependent B cell lymphomas. Nat. Met. 2009, 12, 1369–1376.
- Khan, N.; Kahl, B. Targeting BCL-2 in Hematologic Malignancies. Target. Oncol. 2018, 13, 257–267.
- Davids, M.S.; Roberts, A.W.; Seymour, J.F.; Pagel, J.M.; Kahl, B.S.; Wierda, W.G.; Puvvada, S.; Kipps, T.J.; Anderson, M.A.; Salem, A.H.; et al. Phase I First-in-Human Study of Venetoclax in Patients with Relapsed or Refractory Non-Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 826–833.
- Schuetz, J.M.; Johnson, N.A.; Morin, R.D.; Scott, D.W.; Tan, K.; Ben-Nierah, S.; Boyle, M.J.; Slack, G.W.; Marra, M.A.; Connors, J.M.; et al. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia 2012, 26, 1383–1390.
- Younes, A.; Ansell, S.; Fowler, N.; Wilson, W.; de Vos, S.; Seymour, J.; Advani, R.; Forero, A.; Morschhauser, F.; Kersten, M.J.; et al. The landscape of new drugs in lymphoma. Nat. Rev. Clin. Oncol. 2017, 14, 335–346.
- Roschewski, M.; Staudt, L.M.; Wilson, W.H. Diffuse large B-cell lymphoma—Treatment approaches in the molecular era. Nat. Rev. Clin. Oncol. 2014, 11, 12–23.
- Wilson, W.; O’Connor, O.A.; Czuczman, S.; LaCasce, A.S.; Gerecitano, J.F.; Leonard, J.P.; Tulpule, A.; Dunleavy, K.; Xiong, H.; Chiu, Y.-L.; et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010, 11, 1149–1159.
- Clark, M.C.; Pang, M.; Hsu, D.K.; Liu, F.-T.; de Vos, S.; Gascoyne, R.D.; Said, J.; Baum, L.G. Galectin-3 binds to CD45 on diffuse large B-cell lymphoma cells to regulate susceptibility to cell death. Blood 2012, 120, 4635–4644.
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell 2011, 146, 904–917.
- Amorim, S.; Stathis, A.; Gleeson, M.; Iyengar, S.; Magarotto, V.; Leleu, X.; Morschhauser, F.; Karlin, L.; Broussais, F.; Rezai, K.; et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: A dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016, 3, e196–e204.
- Boi, M.; Gaudio, E.; Bonetti, P.; Kwee, I.; Bernasconi, E.; Tarantelli, C.; Rinaldi, A.; Testoni, M.; Cascione, L.; Ponzoni, M.; et al. The BET Bromodomain inhibitor OTX015 affects pathogenic pathways in preclinical B-cell tumor models and synergizes with targeted drugs. Clin. Cancer. Res. 2015, 21, 1628–1638.
- Grommes, C.; Pastore, A.; Palaskas, N.; Tang, S.S.; Campos, C.; Schartz, D.; Codega, P.; Nichol, D.; Clark, O.; Hsieh, W.-Y.; et al. Ibrutinib Unmasks Critical Role of Bruton Tyrosine Kinase in Primary CNS Lymphoma. Cancer Discov. 2017, 7, 1018–1029.
- Krieg, A.M. CpG Still Rocks! Update on an Accidental Drug. Nucleic Acid Ther. 2012, 22, 77–89.
- Jahrsdorfer, B.; Mühlenhoff, L.; Blackwell, S.E.; Wagner, M.; Poeck, H.; Hartmann, E.; Jox, R.; Giese, T.; Emmerich, B.; Endres, S.; et al. B-Cell Lymphomas Differ in their Responsiveness to CpG Oligodeoxynucleotides. Clin. Cancer Res. 2005, 11, 1490–1499.
- Kelly, P.N.; Romero, D.L.; Yang, Y.; Shaffer, A.L.; Chaudhary, D.; Robinson, S.; Miao, W.; Rui, L.; Westlin, W.F.; Kapeller, R.; et al. Selective interleukin-1 receptor–associated kinase 4 inhibitors for the treatment of autoimmune disorders and lymphoid malignancy. J. Exp. Med. 2015, 212, 2189–2201.
- Scott, J.S.; Degorce, S.L.; Anjum, R.; Culshaw, J.; Davies, R.D.; Davies, N.L.; Dillman, K.S.; Dowling, J.E.; Drew, L.; Ferguson, A.D.; et al. Discovery and Optimization of Pyrrolopyrimidine Inhibitors of Interleukin-1 Receptor Associated Kinase 4 (IRAK4) for the Treatment of Mutant MYD88 L265P Diffuse Large B-Cell Lymphoma. J. Med. Chem. 2017, 60, 10071–10091.
- George, P.; Dasyam, N.; Giunti, G.; Mester, B.; Bauer, E.; Andrews, B.; Perera, T.; Ostapowicz, T.; Frampton, C.; Li, P.; et al. Third-generation anti-CD19 chimeric antigen receptor T-cells incorporating a TLR2 domain for relapsed or refractory B-cell lymphoma: A phase I clinical trial protocol (ENABLE). BMJ Open 2020, 10, e034629.
- Erdmann, T.; Klener, P.; Lynch, J.T.; Grau, M.; Vočková, P.; Molinsky, J.; Tuskova, D.; Hudson, K.; Polanska, U.M.; Grondine, M.; et al. Sensitivity to PI3K and AKT inhibitors is mediated by divergent molecular mechanisms in subtypes of DLBCL. Blood 2017, 130, 310–322.
- Brown, J.R.; Hamadani, M.; Hayslip, J.; Janssens, A.; Wagner-Johnston, N.; Ottmann, O.; Arnason, J.; Tilly, H.; Millenson, M.; Offner, F.; et al. Voxtalisib (XL765) in patients with relapsed or refractory non-Hodgkin lymphoma or chronic lymphocytic leukaemia: An open-label, phase 2 trial. Lancet Haematol. 2018, 5, e170–e180.
- Lenz, G.; Hawkes, E.; Verhoef, G.; Haioun, C.; Lim, S.T.; Heo, D.S.; Ardeshna, K.; Chong, G.; Haaber, J.; Shi, W.; et al. Single-agent activity of phosphatidylinositol 3-kinase inhibition with copanlisib in patients with molecularly defined relapsed or refractory diffuse large B-cell lymphoma. Leukemia 2020, 34, 2184–2197.
- Lam, L.T.; Davis, R.E.; Pierce, J.; Hepperle, M.; Xu, Y.; Hottelet, M.; Nong, Y.; Wen, D.; Adams, J.; Dang, L.; et al. Small molecule inhibitors of IkappaB kinase are selectively toxic for subgroups of diffuse large B-cell lymphoma defined by gene expression profiling. Clin. Cancer Res. 2005, 11, 28–40.
- Strauss, S.J.; Higginbottom, K.; Jüliger, S.; Maharaj, L.; Allen, P.; Schenkein, D.; Lister, T.A.; Joel, S.P. The Proteasome Inhibitor Bortezomib Acts Independently of p53 and Induces Cell Death via Apoptosis and Mitotic Catastrophe in B-Cell Lymphoma Cell Lines. Cancer Res. 2007, 67, 2783–2790.
- Goy, A.; Younes, A.; McLaughlin, P.; Pro, B.; Romaguera, J.E.; Hagemeister, F.; Fayad, L.; Dang, N.H.; Samaniego, F.; Wang, M.; et al. Phase II study of proteasome inhibitor bortezomib in relapsed or refractory B-cell non-Hodgkin’s lymphoma. J. Clin. Oncol. 2005, 23, 667–675.
- Dunleavy, K.; Pittaluga, S.; Czuczman, M.S.; Dave, S.S.; Wright, G.; Grant, N.; Shovlin, M.; Jaffe, E.S.; Janik, J.E.; Staudt, L.M.; et al. Differential efficacy of bortezomib plus chemotherapy within molecular subtypes of diffuse large B-cell lymphoma. Blood 2009, 113, 6069–6076.
- Zhang, L.-H.; Kosek, J.; Wang, M.; Heise, C.; Schafer, P.H.; Chopra, R. Lenalidomide efficacy in activated B-cell-like subtype diffuse large B-cell lymphoma is dependent upon IRF4 and cereblon expression. Br. J. Haematol. 2012, 160, 487–502.
- Ferreri, A.J.M.; Sassone, M.; Angelillo, P.; Zaja, F.; Re, A.; di Rocco, A.; Spina, M.; Fabbri, A.; Stelitano, C.; Frezzato, M.; et al. Long-lasting efficacy and safety of lenalidomide maintenance in patients with relapsed diffuse large B-cell lymphoma who are not eligible for or failed autologous transplantation. Hematol. Oncol. 2020, 38, 257–265.
- Nowakowski, G.S.; LaPlant, B.; Macon, W.R.; Reeder, C.B.; Foran, J.M.; Nelson, G.D.; Thompson, C.A.; Rivera, C.E.; Inwards, D.J.; Micallef, I.N.; et al. Lenalidomide Combined With R-CHOP Overcomes Negative Prognostic Impact of Non–Germinal Center B-Cell Phenotype in Newly Diagnosed Diffuse Large B-Cell Lymphoma: A Phase II Study. J. Clin. Oncol. 2015, 33, 251–257.
- Jin, Z.; Qing, K.; Ouyang, Y.; Liu, Z.; Wang, W.; Li, X.; Xu, Z.; Li, J. Low dose of lenalidmide and PI3K/mTOR inhibitor trigger synergistic cytoxicity in activated B cell-like subtype of diffuse large B cell lymphoma. J. Exp. Clin. Cancer Res. 2016, 35, 1–16.
- Gu, J.J.; Hernandez-Ilizaliturri, F.J.; Kaufman, G.P.; Czuczman, N.M.; Mavis, C.; Skitzki, J.J.; Czuczman, M.S. The novel proteasome inhibitor carfilzomib induces cell cycle arrest, apoptosis and potentiates the anti-tumour activity of chemotherapy in rituximab-resistant lymphoma. Br. J. Haematol. 2013, 162, 657–669.
- Yang, Y.; Shaffer, A.L., III; Emre, N.T.; Ceribelli, M.; Zhang, M.; Wright, G.; Xiao, W.; Powell, J.; Platig, J.; Kohlhammer, H.; et al. Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell 2012, 21, 723–737.
- Mascarenhas, J.; Hoffman, R. Ruxolitinib: The First FDA Approved Therapy for the Treatment of Myelofibrosis: Figure 1. Clin. Cancer Res. 2012, 18, 3008–3014.
- Younes, A.; Romaguera, J.; Fanale, M.; McLaughlin, P.; Hagemeister, F.; Copeland, A.; Neelapu, S.; Kwak, L.; Shah, J.; Faria, S.D.C.; et al. Phase I Study of a Novel Oral Janus Kinase 2 Inhibitor, SB1518, in Patients with Relapsed Lymphoma: Evidence of Clinical and Biologic Activity in Multiple Lymphoma Subtypes. J. Clin. Oncol. 2012, 30, 4161–4167.
- Liu, Y.; Li, P.-K.; Li, C.; Lin, J. Inhibition of STAT3 Signaling Blocks the Anti-apoptotic Activity of IL-6 in Human Liver Cancer Cells. J. Biol. Chem. 2010, 285, 27429–27439.
- Seth, P.P.; Vasquez, G.; Allerson, C.A.; Berdeja, A.; Gaus, H.; Kinberger, G.A.; Prakash, T.P.; Migawa, M.T.; Bhat, B.; Swayze, E.E. Synthesis and biophysical evaluation of 2′,4′-constrained 2′O-methoxyethyl and 2′,4′-constrained 2′O-ethyl nucleic acid analogues. J. Org. Chem. 2010, 75, 1569–1581.
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185.
- Reilley, M.J.; McCoon, P.; Cook, C.; Lyne, P.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: Results of a phase 1b trial. J. Immunother. Cancer 2018, 6, 119.
- Ansell, S.M.; Hurvitz, S.A.; Koenig, P.A.; La Plant, B.R.; Kabat, B.F.; Fernando, D.; Habermann, T.M.; Inwards, D.J.; Verma, M.; Yamada, R.; et al. Phase I Study of Ipilimumab, an Anti–CTLA-4 Monoclonal Antibody, in Patients with Relapsed and Refractory B-Cell Non-Hodgkin Lymphoma. Clin. Cancer Res. 2009, 15, 6446–6453.
- Zinzani, P.L.; Santoro, A.; Gritti, G.; Brice, P.; Barr, P.M.; Kuruvilla, J.; Cunningham, D.; Kline, J.; Johnson, N.A.; Mehta-Shah, N.; et al. Nivolumab Combined with Brentuximab Vedotin for Relapsed/Refractory Primary Mediastinal Large B-Cell Lymphoma: Efficacy and Safety from the Phase II CheckMate 436 Study. J. Clin. Oncol. 2019, 37, 3081–3089.
- Bledsoe, J.R.; Redd, R.A.; Hasserjian, R.P.; Soumerai, J.D.; Nishino, H.T.; Boyer, D.F.; Ferry, J.A.; Zukerberg, L.R.; Harris, N.L.; Abramson, J.S.; et al. The immunophenotypic spectrum of primary mediastinal large B-cell lymphoma reveals prognostic biomarkers associated with outcome. Am. J. Hematol. 2016, 91, E436–E441.
- Roemer, M.G.; Advani, R.H.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J. Clin. Oncol. 2016, 34, 2690–2697.
- Kiyasu, J.; Miyoshi, H.; Hirata, A.; Arakawa, F.; Ichikawa, A.; Niino, D.; Sugita, Y.; Yufu, Y.; Choi, I.; Abe, Y.; et al. Expression of programmed cell death ligand 1 is associated with poor overall survival in patients with diffuse large B-cell lymphoma. Blood 2015, 126, 2193–2201.
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010, 116, 3268–3277.
- Chapuy, B.; Roemer, M.G.M.; Stewart, C.; Tan, Y.; Abo, R.P.; Zhang, L.; Dunford, A.J.; Meredith, D.M.; Thorner, A.R.; Jordanova, E.S.; et al. Targetable genetic features of primary testicular and primary central nervous system lymphomas. Blood 2016, 127, 869–881.
- Armand, P.; Nagler, A.; Weller, E.A.; Devine, S.M.; Avigan, D.E.; Chen, Y.-B.; Kaminski, M.S.; Holland, H.K.; Winter, J.N.; Mason, J.R.; et al. Disabling Immune Tolerance by Programmed Death-1 Blockade with Pidilizumab after Autologous Hematopoietic Stem-Cell Transplantation for Diffuse Large B-Cell Lymphoma: Results of an International Phase II Trial. J. Clin. Oncol. 2013, 31, 4199–4206.
- Zinzani, P.L.; Ribrag, V.; Moskowitz, C.H.; Michot, J.-M.; Kuruvilla, J.; Balakumaran, A.; Zhang, Y.; Chlosta, S.; Shipp, M.A.; Armand, P. Safety and tolerability of pembrolizumab in patients with relapsed/refractory primary mediastinal large B-cell lymphoma. Blood 2017, 130, 267–270.
- Nayak, L.; Iwamoto, F.M.; La Casce, A.; Mukundan, S.; Roemer, M.G.M.; Chapuy, B.; Armand, P.; Rodig, S.J.; Shipp, M.A. PD-1 blockade with nivolumab in relapsed/refractory primary central nervous system and testicular lymphoma. Blood 2017, 129, 3071–3073.
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients with Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704.
- Armand, P.; Engert, A.; Younes, A.; Fanale, M.; Santoro, A.; Zinzani, P.L.; Timmerman, J.M.; Collins, G.P.; Ramchandren, R.; Cohen, J.B.; et al. Nivolumab for Relapsed/Refractory Classic Hodgkin Lymphoma After Failure of Autologous Hematopoietic Cell Transplantation: Extended Follow-Up of the Multicohort Single-Arm Phase II CheckMate 205 Trial. J. Clin. Oncol. 2018, 36, 1428–1439.
- Chang, A.; Schlafer, D.; Flowers, C.R.; Allen, P.B. Investigational PD-1 inhibitors in HL and NHL and biomarkers for predictors of response and outcome. Expert Opin. Investig. Drugs 2018, 27, 55–70.
- Ribrag, V.; Lee, S.T.; Rizzieri, D.; Dyer, M.J.; Fayad, L.; Kurzrock, R.; Andritsos, L.; Bouabdallah, R.; Hayat, A.; Bacon, L.; et al. A Phase 1b Study to Evaluate the Safety and Efficacy of Durvalumab in Combination with Tremelimumab or Danvatirsen in Patients with Relapsed or Refractory Diffuse Large B-Cell Lymphoma. Clin. Lymphoma Myeloma Leuk. 2021, 21, 309–317.e3.