Diffuse large B-cell lymphomas (DLBCL)s, the most common type of Non-Hodgkin’s Lymphoma, constitute a heterogeneous group of disorders including different disease sites, strikingly diverse molecular features and a profound variability in the clinical behavior. Molecular studies and clinical trials have partially revealed the underlying causes for this variability and have made possible the recognition of some molecular variants susceptible of specific therapeutic approaches. The main histogenetic groups include the germinal center, activated B cells, thymic B cells and terminally differentiated B cells, a basic scheme where the large majority of DLBCL cases can be ascribed. The nodal/extranodal origin, specific mutational changes and microenvironment peculiarities provide additional layers of complexity.
1. Introduction
Diffuse large B-cell lymphoma (DLBCL) is one of the most frequent non-Hodgkin’s Lymphoma types. In recent decades, considerable effort has been made to clarify its molecular pathogenesis, which has led to several DLBCL subclasses being identified and specific therapeutic approaches proposed for some of these variants
[1].
The clinical variability of DLBCL is overwhelming; B-cell lymphoma cases with the same morphology (large cells) may originate in the lymph nodes or in any conceivable extranodal localization. They quite often respond to first-line immunochemotherapy, but eventually, some of them relapse and/or progress, with a final 5-year overall survival (OS) probability of around 75%. Standardized protocols for DLBCL staging and treatment have been approved, but in spite of this, the OS and time to progression (TTP) probabilities are still disappointingly low for cases with advanced clinical staging. In contrast to the progress made in the understanding of the molecular basis of these tumors, the therapeutic approach is still mainly based on a combination of immunochemotherapy and cytotoxic therapy
[2].
Molecular studies of DLBCL, using a variety of complementary techniques, have produced a massive amount of information ( and
Supplementary Table S1). DLBCL cells have been shown to carry on multiple combinations of chromosomal translocations involving the
BCL2,
BCL6 and
MYC genes translocated with immunoglobulin heavy or light-chain genes or a myriad of other genes, together with somatic mutations involving several hundred genes regulating the B-cell survival pathways, cell cycle, apoptosis, chromatin conformation, cell metabolism, immune response, DNA repair and others. Combinations of these multiple genetic alterations give rise to a complex scenario in which the identification of precise combinations underlying specific clinicopathological sites of presentation, evolution and response to treatment continues to be a challenge that has so far only been partially addressed.
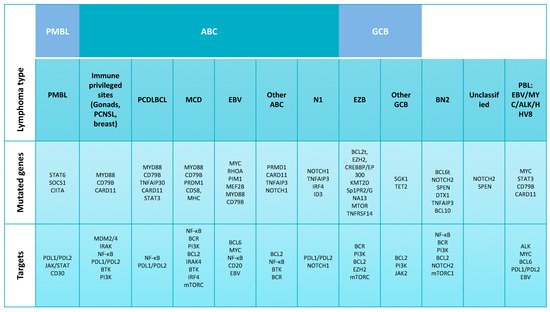
Figure 1. Diffuse large B-cell lymphoma classification and molecular alterations. Subgroups of diffuse large B-cell lymphoma (DLBCL), including its molecular subgroups activated B cell-like (ABC) DLBCL and germinal center B cell-like (GCB) DLBCL defined by gene expression and genetic analysis. Each column represents one subtype. Abbreviations: DLBCL, diffuse large B-cell lymphoma; PTL, primary testicular diffuse large B-cell lymphoma; PCNSL, primary DLBCL of the central nervous system; PMBL, mediastinal large B-cell lymphoma; PCDLBCL, primary cutaneous diffuse large B-cell lymphoma leg-type; MCD, cooccurrence of MYD88L265P and CD79B mutations; EBV, Epstein–Barr virus; N1, NOTCH1 mutations; EZB, EZH2 mutations and BCL2 translocations; BN2, BCL6 fusions and NOTCH2 mutations; PBL, plasmablastic lymphoma; BTK, Bruton’s tyrosine kinase inhibitors; BCR, B-cell receptors; PI3K, phosphoinositide 3-kinase; BCL6t, BCL6 translocation.
Gene expression profiling (GEP) studies led to the identification of different DLBCL molecular subtypes based on the cell of origin (COO) ( and ): germinal center B-cell-like (GCB) and activated B-cell-like (ABC) subtypes
[3][4][3,4]. The COO variability has been found to explain a significant part of the DLBCL molecular heterogeneity
[5][6][5,6], but data concerning its clinical applicability have been controversial
[6][7][8][9][10][11][6,7,8,9,10,11]. The possibility of using COO as a predictor of response to lenalidomide, ibrutinib or bortezomib, when associated with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone), is still contentious
[12][13][14][12,13,14]. Parallel efforts have revealed that a variety of molecular events leading to the deregulation of
MYC and
BCL2 expression, or the simultaneous expression of both protein markers, have also prognostic value, regardless of the COO
[11][15][16][17][18][19][20][21][11,15,16,17,18,19,20,21]. Most of these studies used immunohistochemistry (IHC) to assess the BCL2 and MYC expression, while COO is widely determined using the NanoString platform. Other prognostic markers have been found, including the Epstein–Barr virus (EBV), P53, CD5, CD30, PDL1 and others
[22][23][24][25][26][27][28][22,23,24,25,26,27,28], mostly using IHC or in situ hybridization (ISH) (e.g., EBER) markers.
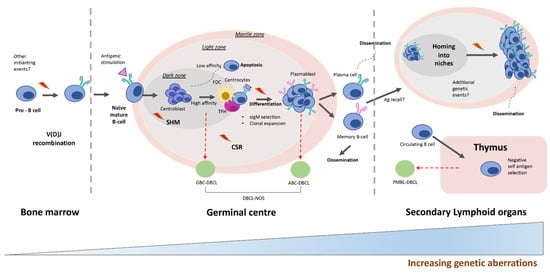
Figure 2. Diffuse large B-cell lymphoma development. Upon stimulation with an antigen (Ag), naive B cells enter the germinal center (GC) reaction, where they undergo rounds of somatic hypermutation (SHM), class-switch recombination (CSR) and proliferation. The initiation of GBC-DLBCL may derive from the transformation of light zone cells. ABC-DLBCL is thought to originate from light zone cells poised to undergo plasma cell differentiation. PMBL is thought to develop from a thymic post-GC B cell or from a GC B cell that has migrated to the thymus. Abbreviations: GBC, germinal center B cell-like; ABC, activated B cell-like; PMBL, mediastinal large B-cell lymphoma; Ag, antigen.
2. Molecular Alterations Defining Aggressive DLBCL
2.1. DLBCL Subclassification
The term “diffuse large B-cell lymphoma” was initially coined to encompass all diffuse B-cell lymphomas with a large cell cytology independently of the organ of origin, molecular history and other prognostic or predictive data. This lymphoma has subsequently been subclassified to recognize the diverse molecular alterations and to integrate the striking differences in survival probability and response to specific targeted therapies. Mediastinal large B-cell lymphoma (PMBL) has emerged as a distinct clinicopathological entity in which different studies coincide in showing that large B-cell lymphomas arising in the mediastinum have a peculiar clinical presentation and histological features, reflecting the underlying characteristic molecular events and bearing important therapeutic implications (). Additionally, GEP studies have confirmed that PMBL is a separate entity from DLBCL at the molecular level and confirmed their similarities with Hodgkin’s lymphoma (HL)
[29]. These results have consolidated PMBL as a distinct entity and have fueled additional efforts to subclassify DLBCL cases.
Besides the attempts to classify DLBCL, the official classification of the World Health Organization (WHO) is based on the work of Alizadeh et al.
[3], which established the ABC, GBC and NOS groups. In addition to these three major subtypes, the WHO recognizes others based on their location or peculiar morphological features
[30]: cutaneous “leg-type” DLBCL (LBCL leg-type)
[31][32][31,32], primary testicular diffuse large B-cell lymphoma (PTL), primary DLBCL of the central nervous system (PCNSL)
[33], T-cell/histiocyte-rich large B-cell lymphoma (THRLBCL)
[34] and EBV-positive DLBCL
[35] ().
In recent years, some authors have divided DLBCL into several subgroups on the basis of additional GEP findings. Monti et al.
[34] defined three groups: “B-cell receptor/proliferation”, “oxidative phosphorylation” and “host response” (HR). The B-cell receptor/proliferation group contains
BCL6 translocations. The oxidative phosphorylation group includes tumors carrying t(14;18) and apoptotic pathway defects. The HR group is characterized by a T-cell and dendritic signature.
The integration of exome sequencing data, copy number analysis and structural variants enabled Chapuy et al. to identify five DBCL subsets (
Supplementary Table S1)
[36], each of which possesses specific genetic features: (C1), low-risk ABC-DLBCLs of extrafollicular/marginal zone origin; (C2), tumors with biallelic inactivation of
TP53, 9p21.3/CDKN2A and associated genomic instability; (C3), high-risk GCB-DLBCLs with
BCL2 structural variants and alterations of the PTEN and epigenetic enzymes; (C4), low-risk GCB-DLBCLs with alterations in the B-cell receptor (BCR)/PI3K, JAK/STAT and BRAF pathway components and multiple histones; (C5), tumors containing
18q gain, including frequent
BCL2 gain.
More recently, Schmitz et al. classified DLBCL into four groups, according to the presence of genetics aberrations
[37]: MCD (cooccurrence of
MYD88L265P and
CD79B mutations), BN2 (
BCL6 fusions and
NOTCH2 mutations), N1 (
NOTCH1 mutations) and EZB (
EZH2 mutations and
BCL2 translocations). Lacy et al. established five molecular subtypes, termed MYD88, BCL2, SOCS1/SGK1, TET2/SGK1 and NOTCH2, also based on the study of genetic alterations, in a cohort of 928 patients ()
[38].
It is of note that some subgroups of DLBCL cases arising in extranodal localizations reproduce the molecular alterations and phenotype that define the MCD subgroup. PCNSL has a particular molecular profile characterized by a predominance of the ABC subtype and the presence of
MYD88/CD79B mutations and PD-1/PD-2 pathway alterations
[39][40][39,40]. Similar observations have been made for cutaneous LBCL leg-type and DLBCL arising in immune-privileged sites such as the testis and breast, among others, and which resemble the ABC subtype
[41], with 60% of mutations occurring in
MYD88L265P [42] and 20% in
CD79B [43].
2.2. Relevant Genes and Pathways
The pathogenesis of DBCL is a good example of a multistep process involving the accumulation of genetic alterations, including somatic mutations, copy number changes, chromosomal translocations and epigenetic changes ( and
Supplementary Table S1)
[44][45][46][47][44,45,46,47]. These changes are closely linked to two main physiological mechanisms that operate during the immunoglobulin (IG) DNA remodeling processes in B lymphocytes: chromosomal translocations, which arise from errors in V(D)J recombination, somatic hypermutation (SHM) and immunoglobulin class-switch recombination (CSR)
[46] () and activation-induced cytidine deaminase (AID)-mediated SHM
[47]. Additionally, different B-cell lymphoma genetic alterations affect the crosstalk between malignant B cells and the surrounding cells, including follicular dendritic cells and follicular helper T cells
[48].
The introduction of next-generation sequencing (NGS) technologies and functional genomic analysis has revealed an unexpectedly high degree of diversity of the mutational landscape of DBCL. Somatic mutations have been identified in more than 700 genes
[44] (
Supplementary Table S1), with an average of 50–100 alterations in the coding regions per case (including mutations and copy number alterations)
[36][37][49][50][36,37,49,50]. Around 150 of these genes are mutated driver genes, including some that occur at low frequencies
[51]. Notably, Chapuy et al. showed that 80% of the observed mutations are associated with the spontaneous deamination of cytosines at CpGs and involve a switch from cytosine to thymine (C > T)
[52].
3. B-Cell Receptor Signaling and Toll-Like Receptor Pathways
3.1. BCR
During the GC reaction, T-follicular helper (T
FH) cells positively select only centrocytes whose BCRs have a high affinity for antigen(s) so that they can enter the CSR process, having been activated by AID (). The BCR is a transmembrane signaling complex composed of an antigen recognition unit and a signaling unit (). The signaling unit comprises a heterodimer of CD79A and CD79B proteins and transduces the signal to a gene complex (denominated by the My-T-BCR supercomplex)
[53][80] once the BCR has recognized the antigen. This signal ultimately regulates B-cell survival. The pathway is recurrently deregulated as a consequence of somatic mutations. These pathway alterations are much more frequent in cases of the DLBCL-ABC type that depend on BCR signaling. Thus, mutations targeting the immunoreceptor tyrosine-based activation motifs (ITAMs) in
CD79A and
CD79B are present in ~20% of ABC patients but only in ~3% of GBC patients
[54][81]. ABC cases have been shown to carry PRDM1-truncating mutations (~20%) and homozygous deletions (~4%)
[37][55][37,82].
PRDM1 is one of the key genes involved in regulating the BCR pathway, wherein its function is to inhibit BCR signaling
[56][83]. CARD11 encodes a scaffold protein that, following its activation by PKCβ, recruits BCL-10 and MALT1 to activate the JNK pathway
[54][81]. Mutations in
CARD11 result in a gain-of-function phenotype that activates the NF-κB pathway
[57][84]. On the other hand, BCL-10 is overexpressed in ~25% of GBC and ~11% of ABC cases, mostly due to the presence of translocations
[58][85]. Most potential pathogenic mutations in
BCL10 are located in the carboxy-terminal domain. These also affect both subtypes: ~10% of ABC and ~6% of GBC cases
[37]. Amplifications in MALT1 are mainly detected in ABC-DLBCL (~7% of cases) and GBC (~1%)
[37]. These multiple mutations interact to induce lymphomagenic CARD11/BCL10/MALT1 signaling, which drives malignant B-cell proliferation via cooperative NF-κB and JNK activation
[59][86].
3.2. Toll-Like Receptor Signaling
MYD88 is a signal adaptor protein that mediates the activation of the NF-κB pathway after the stimulation of the Toll-like receptor (TLR) and the interleukin IL-1 and IL-18 receptors ()
[60][61][87,88]. MYD88 is frequently activated in DLBCL-ABC and other B-cell lymphoma types, most often as a consequence of a redundant L265P mutation
[37][62][37,89]. The
MYD88L265P mutation, located in the Toll/IL-1 receptor domain of MYD88, intensifies the interaction and consecutive phosphorylation of the IRAK1 and IRAK4 complex, activating downstream targets, including NF-κB and JAK–STAT signaling
[60][87]. A subset of DLBCLs presents concomitant
MYD88L265Pmutations and
CD79B or
CD79A (34% of the cases with a
MYD88L265P mutation had a coincident
CD79B/A mutation; these are termed MCD) ()
[60][87], providing evidence of the cooperative role of these mutations in the ABC subgroup pathogenesis.
3.3. NF-κB Pathway
NF-κB is a transcription regulator that, after activation by various intra- and extracellular stimuli, translocates to the nucleus and stimulates the expression of the genes involved in a wide variety of biological functions, including cell growth and apoptosis inhibition (). The inappropriate activation of NF-κB is associated with several inflammatory and lymphoproliferative disorders. The raised level of expression of an NF-κB signature is a specific defining feature of the ABC subtype
[63][90]. The ABC-DLBCL subtype shows a constitutive activation of the NF-κB signaling cascade as a consequence of the genetic alterations in NF-κB modifiers and/or EBV infection
[63][90]. Ultimately, genetic abnormalities that activate the BCR and TLR pathways bring about NF-κB activation. For example, the mutations in
CARD11 enhance its capacity to transactivate the NF-κB genes
[64][91], while inactivating mutations of the negative regulator
TNFAIP3, found in ~30% of the cases, may cause an increase of the NF-κB response and subsequent neoplastic transformation
[65][66][92,93].
TNFAIP3, which is more frequent in the BN2 group, cooccurs with
MYD88L265P mutations in around 7% of ABC patients, suggesting that they may cooperate in ABCL pathogenesis
[67][68][94,95].
3.4. PI3K/AKT/mTOR Pathway
The PI3K signaling pathway is located downstream of BCR signaling and is activated by CD19 and the SYK kinase ()
[69][96]. PI3K activates AKT, which sends the signal to mTOR and the other signaling pathways
[70][97]. Genetic aberrations are present in several genes of the PI3K pathway genes (
RHOA,
GNA13 and
SGK1) in around 34% of DLBCLs
[37]. PTEN is a negative regulator of PI3K signaling, and mutational deletions in the
PTEN gene facilitate activation of the PI3K/AKT pathway
[71][98]. The microRNA
MIR17HG that targets the mRNA of
PTEN is amplified mostly commonly in GCB-DLBCLs (~8% of cases)
[37], leading to a reduced PTEN expression. Mutations in PI3K itself occur only in ABC cases (6%)
[37]. Furthermore, mutations in the PI3K/AKT pathway can indirectly activate the NF-κB pathway, causing a malignant transformation
[72][99].
FOXO1 is a transcription factor that acts as a tumor suppressor. It is phosphorylated by AKT, resulting in cytoplasmic sequestration and suppression of its activity
[73][100]. Mutations in
FOXO1 have been observed in around 8% of DLBCL cases
[74][75][65,101] and exhibit aberrant nuclear localization
[75][101].
4. Therapeutic Targeting of Diffuse Large B-Cell Lymphoma
Molecular analysis and clinical trials in DLBCL are progressively revealing multiple therapeutic opportunities that eventually could replace the currently used chemotherapy. These new options are tumor-type specific and, thus, adapted for the GC, ABC, thymic or terminally differentiated phenotypes and, additionally, in many cases, are coupled with precise genetic events or deregulated pathways.
4.1. BCL6 Inhibitors
The
BCL6-deregulated expression is central to DLBCL and FL molecular pathogenesis and makes the molecule an attractive therapeutic target. Although the direct targeting of BCL6 is difficult, small molecule inhibitors have been generated that bind to BCL6 and block corepressor recruitment
[76][77][146,147]. These are active in ABC- and GC-DLBCL cases.
4.2. BCL2/MYC Inhibitors
An increased BCL2 expression as a consequence of the
BCL2 gene translocation or the oncogenic activation of multiple cell survival pathways is a frequent finding in DLBCL, especially in the ABC subtype, making this molecule an ideal target for therapy
[78][148].
Venetoclax is a BCL2 inhibitor currently under investigation for the treatment of DLBCL
[79][149], following its approval for use against chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL). It targets the BH3 domain, where most BCL2 mutations occur
[80][79]. The use of venetoclax in combination with chemotherapy, including R-CHOP and R-EPOCH, is also being studied for DHL and double-expresser DLBCL
[81][82][150,151]. Navitoclax is a BCL2 inhibitor resembling BH3 that has a proven activity in CLL and NHL patients. However, the clinical application of this drug is limited due to its dose-dependent thrombocytopenia effect
[83][152].
Gal-3 inhibitors interrupted CD45/Gal-3 interaction and restored apoptotic function in their preclinical models
[84][153].
MYC is under the epigenetic regulation of bromodomain (BRD)-containing proteins that recruit transcription factors to acetylate chromatin, leading to gene transcription
[85][154]. A range of BRD inhibitors have demonstrated some degree of clinical activity in phase I trials
[86][87][155,156]. However, in spite of these promising findings, much more experimental work and further clinical trials need to be done to explore the possibilities of MYC silencing using bromodomain and extraterminal (BET) inhibitors.
4.3. BTK iInhibition
B-cell receptor signaling has emerged as a key survival factor for normal and neoplastic B-lymphocytes. Thus, the cell viability of large B-cell lymphomas with an ABC phenotype has been shown to depend on NF-κB activation via chronic active BCR signaling
[54][81] through the formation of a complex including
MYD88L265P with IRAK kinases that activates NF-κB and JAK-STAT signaling
[60][87]. BTK inhibitors have been developed and tested in diverse experimental and clinical studies, which showed that the tumors with the MCD genetic subtype (
CD79B and
MYD88L265P) had a particularly high response rate
[18]. Thus, although the ABC phenotype has not been shown to predict the responses to lenalidomide, ibrutinib or bortezomib
[12][13][14][18][12,13,14,18], a subgroup of these cases defined by the presence of mutations in
CD79B and
MYD88L265P seems to have identified a group of DLBCL cases that may respond to ibrutinib
[18], although confirmation of this would require a clinical trial designed specifically for this purpose. This positive result in a subset of the ABC subtype also provides additional useful information about PCNSLs, which are tumors with an ABC phenotype (and frequent
CD79B and
MYD88L265P mutations) that are difficult to treat. Clinical responses to ibrutinib have been noted in 77% of patients with PCNSL, five cases of which were complete responses
[88][157].
4.4. Toll-Like Receptor Inhibition
ABC-DLBCL bear mutations, copy number alterations and amplifications in the TLR components involving
MYD88,
TLR9,
CNPY3 and
UNC93B1 [53][80]. Of these, TLR9 has been selected as a therapeutic target in two clinical trials, although it proved to be of limited efficacy
[89][90][158,159]. MYD88 is an adaptor protein that mediates Toll and IL-1 receptor signaling
[61][88]. Notably, RNAi experiments have revealed that MYD88 and the associated IRAK1 and IRAK4 are indispensable for ABC-DLBCL survival
[60][87]. Due to the great relevance of MYD88 in DLBCL pathogenesis, several inhibitors of IRK4 have been proposed, and experimental studies have shown the pharmacological inhibition of IRAK4 to be a suitable therapeutic strategy for treating ABC-DLBCL, especially in combination with the BTK inhibitor ibrutinib or the Bcl-2 inhibitor ABT-199
[91][92][160,161].
A phase I trial study will analyze a third generation of anti-CD19 chimeric antigen receptor T cells, incorporating a TLR2 domain in patients with relapsed or refractory (R/R) B-cell lymphoma
[93][162].
4.5. PI3K Inhibition
Experimental studies have demonstrated that GC and ABC-type DLBCLs are both sensitive to PI3K/AKT inhibition, although for different reasons
[94][163]. AKT signaling is known to be crucial for PTEN-deficient DLBCLs, whereas the PI3Kα/δ-induced activation of NF-κB is critical for ABC-DLBCLs
[94][163]. Clinical trials have yielded interesting results in DLBCLs treated with PI3K/AKT inhibitors. For example, voxtalisib (also known as XL765 or SAR245409), a pan-PI3K/mTOR inhibitor, in patients with relapsed or refractory DLBCL is only slightly clinically active
[95][164], but copanlisib (a PI3K inhibitor with potent activity against the PI3K-α and -δ isoforms) has a higher response rate in ABC-DLBCL than in GCB-DLBCL patients
[96][165].
4.6. NF-κB Inhibition
Several therapeutic strategies employ the NF-κB pathway as a target, mainly in ABC-DLBCL. Small-molecule inhibitors of the IκB kinase (IKK) complex have demonstrated selective inhibition of the ABC-DLBCL cell lines
[97][166]. On the other hand, bortezomib targets the NF-κB pathway through reversible proteasome inhibition and by blocking the degradation of the NF-κB inhibitory protein IκBα
[98][167]. Bortezomib in patients with R/R DLBCL has a lower efficacy when administered alone than when combined with the EPOCH regimen
[99][100][168,169].
Lenalidomide is a well-known drug that blocks the BCR–NF-κB pathway by targeting the E3 ubiquitin ligase component cereblon, with antineoplastic consequences
[101][170]. The efficacy of lenalidomide maintenance has yielded positive results in DLBCL patients after salvage or frontline therapy
[102][171]. DLBCL demonstrated a substantial activity in patients with R/R, especially in the non-GBC and ABC-DLBCL subtypes. In newly diagnosed DLBCL patients, the administration of lenalidomide with CHOP seems to have positive effects, particularly in non-GBC patients
[103][172]. Additionally, the combination of lenalidomide with a PI3K inhibitor and mTOR in ABC-DLBCL cells had a synergistic cytotoxic effect
[104][173]. The benefits of lenalidomide in combination with R-CHOP, and alone in ABC cases, merit further investigation. Carfilzomib is a second-generation proteasome inhibitor that has shown promising results in DLBCL cell lines, including those resistant to rituximab
[105][174].
The NF-κB pathway can be indirectly dysregulated by genetic alterations in other pathways, such as alterations of the BCR signaling pathway components
[54][64][81,91]. Additionally, mutations in the adaptor molecule MYD88, which are present in approximately 30% of ABC cases, might alter the signal transduction from the TLR to the NF-κB complex
[60][87]. In a clinical trial study, tumors with concomitant mutations in
MYD88 and
CD79B responded well to BTK inhibition treatment, unlike those carrying only
MYD88 mutations
[18].
4.7. JAK/STAT Inhibition
The JAK/STAT signaling cascade transduces signals to the nucleus, where they regulate key biological functions such as proliferation and cell survival. Recurrent genetic alterations in this pathway are present in DLBCL, making it an interesting therapeutic target
[106][175].
Ruxolitinib is an oral inhibitor of JAK1 and JAK2 that has been approved for the treatment of primary myelofibrosis
[107][176]. It is currently being investigated in the context of R/R DLBCL, although it seems to be more effective in combination regimens. Pacritinib is an oral small-molecule inhibitor that selectively inhibits JAK2 and has shown efficacy in vitro in DLBCL cell lines
[108][177]. It is of note that the constitutive activation of STAT3 is associated with an aggressive disease phenotype and poor overall survival
[109][178]. AZD9150 is a 16-nucleotide next-generation chemistry antisense oligonucleotide
[110][179] that targets STAT3 mRNA and downregulates its expression. Preclinical and phase 1b trial studies have demonstrated its efficacy and safety in patients with refractory/resistant DLBCL
[111][112][180,181].
4.8. ICIs
Immune checkpoint inhibitors (ICIs) blockading CTLA4, PD1 and PD-L1 have an indisputable role in the treatment of PMBCL and HL
[113][114][182,183] and in a subset of DLBCL cases.
PDL1 and PDL2 are both frequently expressed in PMBL (~71% of cases)
[115][184], HL (~97%)
[116][185] and ABC-DLCBCL (~36% PDL1 and ~60% PDL2) and GBC-DLBCL (~4% PDL1 and ~26% PDL2)
[117][186]. The causal mechanisms of PDL1 and/or PDL2 overexpression include amplification of the 9p24 genomic area, where PDL1 and PDL2 are located, and Epstein–Barr virus infection. Although the alteration of 9p24 occurs in most of the PMBL patients
[118][129], it is not restricted to PMBL, since it has also been described in HL
[116][185] in around 54% of PTL and 52% of PCNSL cases
[119][187], as well as in a subset of DLBCL (19%) cases with refractory/relapsed DLBCL
[120][121][188,189]. Consistent with these observations, the PD-1 blockade with nivolumab is clinically active in primary CNS and testicular lymphoma
[122][190].
The potential therapeutic activity of the anti-PD1 antibody (nivolumab and others) has been evaluated in a small number of DLBCL patients
[123][191] who showed a lower response rate (40%) than seen in HL patients (66%)
[124][192]. Preliminary studies of other antibodies involving PDL1, e.g., atezolizumab, have yielded promising results
[125][126][193,194]. Studies of therapies involving PD-L1 antibodies in combination with other antibodies against different immune checkpoints, or in combination with chemotherapy, are currently being conducted.
A number of clinical trials are also currently assessing the safety and efficacy of different antibodies that target other immune checkpoints, such as varlilumab, which targets the tumor necrosis factor receptor superfamily member 9 (4-1BB).