In an ageing society, polypharmacy has become a major public health and economic issue. Overuse of medications, especially in patients with chronic diseases, carries major health risks. One common consequence of polypharmacy is the increased emergence of adverse drug events, mainly from drug–drug interactions. The majority of currently available drugs are metabolized by CYP450 enzymes. Interactions due to shared CYP450-mediated metabolic pathways for two or more drugs are frequent, especially through reversible or irreversible CYP450 inhibition. The magnitude of these interactions depends on several factors, including varying affinity and concentration of substrates, time delay between the administration of the drugs, and mechanisms of CYP450 inhibition. Various types of CYP450 inhibition (competitive, non-competitive, mechanism-based) have been observed clinically, and interactions of these types require a distinct clinical management strategy.
- keyword
- CYP450
- Drug-drug interactions
- mechanism based inhibition
1. Introduction
Inhibition of cytochrome P450 (CYP450) enzymes is the most common mechanism leading to drug–drug interactions [4][1]. CYP450 inhibition can be categorized as reversible (including competitive and non-competitive inhibition) or irreversible (or quasi-irreversible), such as mechanism-based inhibition. Each type of interaction involves a distinct clinical management strategy. This is why a comprehensive understanding of mechanisms of CYP450-mediated metabolism inhibition is needed to prevent or mitigate these harmful drug interactions.
2. Mechanism of CYP450 Inhibition
Drug interactions due to drug metabolism inhibition are frequent since (1) CYP450-mediated metabolism is the major route of elimination for a large number of drugs, and (2) multiple drugs can compete for the same CYP450 active site. Mechanisms of CYP450 inhibition can be categorized as reversible (including competitive or non-competitive) or irreversible/quasi-irreversible (mechanism-based inhibition).
2.1. Reversible CYP450 Inhibition
Reversible inhibition is a result of rapid association and dissociation between the substrate drugs and the enzyme and can be categorized as competitive or non-competitive. A third category, uncompetitive inhibitor, also considered as a reversible inhibition type, is a very rare phenomenon and will not be considered in this current review; this type of inhibitor binds only the enzyme–substrate complex, leading to a dead-end complex.
2.1.1. Competitive Inhibition
The ability of a single CYP450 isoform to metabolize multiple substrates is responsible for several drug interactions associated with reversible competitive inhibition. Competitive inhibition occurs when two substrates compete for the same active site—such as the prosthetic heme iron or substrate-binding region—of CYP450s. The competition is a function of the respective affinities of the two substrates for the binding site and their concentrations in the proximity of the enzyme. First, the most clinically relevant situation will be discussed.
-
Two Substrates with Different Affinities Administered Concomitantly
This situation is often encountered in clinical practice. Under a competitive inhibition condition, a substrate with strong affinity (acting as a perpetrator) can displace a weaker substrate (behaving as a victim) from the active site (Figure 1), thus increasing the Km of the victim drug (decreased affinity) and reducing the extent of its breakdown (decrease in its CLint) over a period of time.
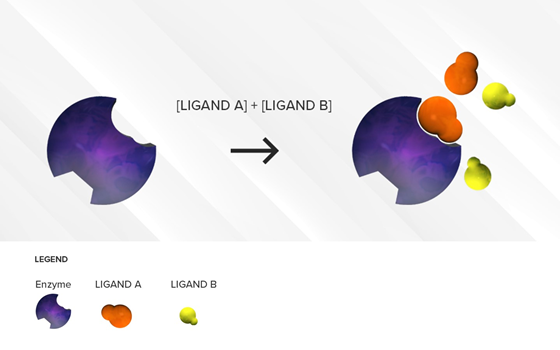
Figure 1. Illustration of reversible competitive inhibition where ligand A (orange) is a substrate with strong affinity and ligand B (yellow) is a substrate with a weaker affinity for a specific enzyme (purple). As long as the concentrations of the two substrates are comparable, the stronger affinity substrate with higher binding affinity will be preferred at the active site of the enzyme resulting in an accumulation of ligand B.
-
Two Substrates with Largely Different Concentrations
As mentioned previously, the competitive inhibition process is sensitive to substrate concentrations. If concentrations of the weaker affinity substrate are much higher than concentrations of the stronger affinity substrate, the weaker affinity substrate can displace the stronger affinity substrate and overcome the enzyme inhibition, which is why this type of inhibition is deemed reversible. (Figure 2) The greater the difference there is between the affinity of the weaker affinity substrate and the stronger affinity substrate, the more the concentration of the weaker affinity substrate needs to be increased to displace the stronger affinity substrate. This situation can be observed clinically when very high concentrations of a weak affinity substrate are present in the intestine or liver (high micromolar concentrations), following its oral administration, while concentrations of another higher affinity substrate have long been absorbed and distributed to various tissues leading to plasma concentrations in the low nanomolar range. In this case, the extent of victim drug inhibition would be minimal. A direct application of this principle is to alleviate the degree of inhibition by separating the time of administration of the two competing drugs.
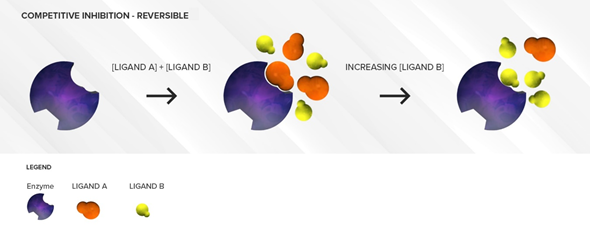
Figure 2. Illustration of reversible competitive inhibition where ligand A (orange) is a substrate with strong affinity and ligand B (yellow) is a substrate with weaker affinity for a specific enzyme (purple). When the concentrations of the weaker affinity substrate are sufficiently high, it can outcompete the stronger affinity substrate for the active site of the enzyme.
2.1.2. Non-Competitive Inhibition
The non-competitive inhibitor does not generally have any structural resemblance to the substrate as it binds to an allosteric site. The non-competitive inhibitor will cause a conformational change in the structure of the active site such that the active site loses its affinity for the substrate. (Figure 3) Thus, there is no direct competition between the inhibitor and the substrate at the active site. This type of inhibition is often long lasting and cannot be overcome by increasing substrate concentrations. Under these conditions, a decrease in the CLint of the substrate due to a decrease in its Vmax is observed. Similar to competitive inhibitors, non-competitive inhibitors also have an almost immediate effect. As long as the concentration of the inhibitor is not changed, the amount of inhibition will not increase over time. This type of inhibition does not require the involvement of NADPH as a cofactor, i.e., the inhibitor is not metabolized by the enzyme, but merely sits in an allosteric site. Other non-competitive inhibition conditions may involve CYPb5 and/or CYP450 oxidoreductase as these factors have been shown to modulate CYP450 activities, at least in in vitro systems [13,14][2][3]. Separating the time of dosing will not alleviate non-competitive inhibition. Fluvoxamine (CYP2C19) and terbinafine (CYP2D6) are some common examples of non-competitive inhibitors at other CYP isoforms [15–17][4][5][6].
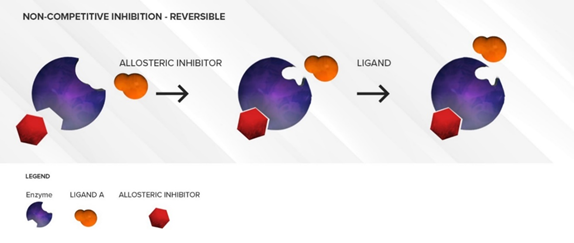
Figure 3. Illustration of reversible non-competitive inhibition. An inhibitor (red) binds to an allosteric site on the enzyme and causes conformational changes that prevent a substrate (orange) from binding to the active site. Over time, as the inhibitor is flushed out, the conformation of the enzyme can return to normal and substrate (orange) can bind to the active site again.
2.1.3. Mixed inhibition
In the case of mixed inhibition, both competitive and non-competitive inhibition occur. Mixed inhibitors can simultaneously bind to both the heme iron atom (at the active site) and lipophilic regions of the protein (allosteric site). Mixed inhibitors are usually more potent inhibitors than competitive or non-competitive inhibitors. Ketoconazole and fluconazole, both imidazole antifungals, exhibit potent mixed reversible inhibition of CYP3As. However, fluconazole is a weaker mixed reversible inhibitor compared to ketoconazole, mainly due to its lower lipophilicity (less binding to an allosteric site).
For a CYP3A substrate like midazolam, concomitant use of non-competitive or mixed CYP3A inhibitors will reduce its transformation to α-hydroxy midazolam, increasing midazolam plasma levels and augmenting the risk of adverse drug events [18][7].
2.2. Irreversible CYP450 Inhibition
Several clinically important pharmacokinetic drug interactions result from a decrease in the metabolic clearance of a substrate due to CYP450 irreversible inhibition. Mechanism-based inhibition is a condition often encountered with irreversible CYP450 inhibitors.
2.2.1 Mechanism-based inhibition
Mechanism-based inhibition can be irreversible or quasi-irreversible. It generally derives from the activation of a substrate drug by a CYP450 isoform into a reactive metabolite, which binds to the enzyme heme prosthetic site (part of the active site), resulting in irreversible long-lasting loss of enzyme activity (decrease in Vmax). (Figure 4) Several drugs undergo metabolic activation by a specific CYP450 isoform to produce inhibitory intermediate metabolites, which can form stable intermediate complexes. As a result, the CYP450 isoform is sequestered in an inactive state. Even though the reactive intermediate metabolite plays a key role in the mechanism-based inactivation of the CYP450 isoform, in many instances, the exact reactive metabolite involved in this phenomenon is unknown.

Figure 4. Illustration of mechanism-based inhibition. The mechanism-based inhibitor (orange) binds to the active site as a substrate. During the normal process of metabolism, it forms either stable intermediate–enzyme complexes or reactive electrophilic species that can lock up or destroy the enzyme, and new enzyme synthesis is required to restore the enzymatic activity.
In the case of quasi-irreversible inhibition, the metabolites form very stable complexes with the heme prosthetic site (metabolite–intermediate complex), so that the enzyme is sequestered in a functionally inactive state. This phenomenon is called quasi-irreversible since, in theory, this complex can be disrupted. In the case of irreversible inhibition, the metabolites covalently bind to the heme prosthetic site or the protein part of the CYP450, leading to irreversible inactivation [19,20][8][9].
Hence, mechanism-based inhibition is active site mediated, and the allosteric site is not involved. In contrast to reversible inhibition mechanisms, mechanism-based inhibition is time dependent and NADPH dependent. This means that the enzyme has to start breaking down the substrate in order for inhibition to proceed. As more drug molecules are metabolized, more complexes are stably formed in the active sites, increasing inhibition over time before it reaches a plateau. Mechanism-based inhibition is therefore also saturable. New enzyme formation is necessary to restore activity: the relationship between the amount of intermediate complex formed and the speed of new enzyme synthesis dictate the equilibrium and extent of enzyme inhibition.
Mechanism-based inhibitors can be classified into two categories: metabolic–intermediate complex formation inhibitors and protein and/or heme alkylation inhibitors.
-
Metabolic–Intermediate Complex Formation (or Alternate Substrate Inhibition)
Such a condition occurs when a stable intermediate metabolite formed during the normal metabolic cycle forms covalent bonds at the active site. This stable intermediate–enzyme complex is not easily broken by increasing substrate concentration. Since the enzyme structure remains otherwise unchanged, theoretically this reaction is reversible with time. However, in in vivo conditions, with this metabolic intermediate complex being excessively stable, the metabolic intermediate cannot be displaced and the enzyme remains inaccessible for metabolism so the reaction seems irreversible.
An example of alternate substrate inhibition is observed with paroxetine as its methoxy diene carbon moiety was found to be responsible for the formation of covalent bonds at the active site of CYP2D6 [21,22][10][11]. Another example of this type of inhibition was observed with clarithromycin when the nitrosoalkene intermediate generated by N-demethylation forms covalent bonds with the active site of CYP3A4 [23][12].
-
Protein and/or Heme Alkylation (or Suicide Inhibition)
This situation takes place when a latent highly reactive (generally electrophilic) intermediate is formed in the catalysis process. The reactive intermediate forms covalent bonds (strong irreversible bonds) with the enzyme in a step that is not part of the normal metabolic pathway. This process can change the conformational structure of the enzyme significantly—it can even destroy the enzyme in some cases—making it functionally unviable. For example, inhibition of CYP2C19 by esomeprazole was found to be mediated by crosslinking the heme and apoprotein moieties in the enzyme, changing its conformational structure [24][13].
It is important to note that since mechanism-based inhibitors are substrates of the enzyme, they can also cause acute competitive inhibition when co-administered with other sensitive substrates. The difference between competitive inhibition and mechanism-based inhibition is that as the time period of exposure to mechanism-based inhibitors increases, the degree of inhibition also increases. (Table 1)
Table 1. Summary of major pharmacokinetic characteristics of various drug inhibition models.
|
Characteristics |
Inhibitor Type |
||
|
Competitive |
Non-Competitive Non-Mechanism Based |
Mechanism-Based |
|
|
Metabolism required |
No |
No |
Yes |
|
Active site mediated |
Yes |
No |
Yes |
|
Time dependent |
No |
No |
Yes |
|
Substrate concentration dependent |
Yes |
No |
Yes |
|
Km (victim drug) |
↑ |
« |
« |
|
Vmax (victim drug) |
« |
¯ |
¯ |
|
CLint (victim drug) |
¯ |
¯ |
¯ |
References
- Lynch, T.; Price, A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am. Fam. Physician 2007, 76, 391–396.
- Zhang, H.; Gao, N.; Liu, T.; Fang, Y.; Qi, B.; Wen, Q.; Zhou, J.; Jia, L.; Qiao, H. Effect of Cytochrome b5 Content on the Activity of Polymorphic CYP1A2, 2B6, and 2E1 in Human Liver Microsomes. PLoS ONE 2015, 10, e0128547.
- Bart, A.G.; Scott, E.E. Structural and functional effects of cytochrome b(5) interactions with human cytochrome P450 enzymes. J. Biol. Chem. 2017, 292, 20818–20833.
- Abdel-Rahman, S.M.; Gotschall, R.R.; Kauffman, R.E.; Leeder, J.S.; Kearns, G.L. Investigation of terbinafine as a CYP2D6 inhibitor in vivo. Clin. Pharmacol. Ther. 1999, 65, 465–472.
- Vickers, A.E.M.; Sinclair, J.R.; Zollinger, M.; Heitz, F.; Glänzel, U.; Johanson, L.; Fischer, V. Multiple Cytochrome P-450s Involved in the Metabolism of Terbinafine Suggest a Limited Potential for Drug-Drug Interactions. Drug Metab. Dispos. 1999, 27, 1029–1038.
- Rasmussen, B.B.; Nielsen, T.L.; Brøsen, K. Fluvoxamine inhibits the CYP2C19-catalysed metabolism of proguanil in vitro. Eur. J. Clin. Pharmacol. 1998, 54, 735–740.
- Lam, Y.W.; Alfaro, C.L.; Ereshefsky, L.; Miller, M. Pharmacokinetic and pharmacodynamic interactions of oral midazolam with ketoconazole, fluoxetine, fluvoxamine, and nefazodone. J. Clin. Pharmacol. 2003, 43, 1274–1282.
- Drolet, B.; Khalifa, M.; Daleau, P.; Hamelin, B.A.; Turgeon, J. Block of the rapid component of the delayed rectifier potassium current by the prokinetic agent cisapride underlies drug-related lengthening of the QT interval. Circulation 1998, 97, 204–210.
- Naritomi, Y.; Teramura, Y.; Terashita, S.; Kagayama, A. Utility of microtiter plate assays for human cytochrome P450 inhibition studies in drug discovery: Application of simple method for detecting quasi-irreversible and irreversible inhibitors. Drug Metab. Pharmacokinet. 2004, 19, 55–61.
- Uttamsingh, V.; Gallegos, R.; Liu, J.F.; Harbeson, S.L.; Bridson, G.W.; Cheng, C.; Wells, D.S.; Graham, P.B.; Zelle, R.; Tung, R. Altering Metabolic Profiles of Drugs by Precision Deuteration: Reducing Mechanism-Based Inhibition of CYP2D6 by Paroxetine. J. Pharmacol. Exp. Ther. 2015, 354, 43–54.
- Bertelsen, K.M.; Venkatakrishnan, K.; Von Moltke, L.L.; Obach, R.S.; Greenblatt, D.J. Apparent mechanism-based inhibition of human CYP2D6 in vitro by paroxetine: Comparison with fluoxetine and quinidine. Drug Metab. Dispos. 2003, 31, 289–293.
- Kouladjian, L.; Chen, T.F.; Gnjidic, D.; Hilmer, S.N. Education and Assessment of Pharmacists on the Use of the Drug Burden Index in Older Adults Using a Continuing Professional Development Education Method. Am. J. Pharm. Educ. 2016, 80, 63.
- Ogilvie, B.W. An In Vitro Investigation into the Mechanism of the Clinically Relevant Drug-Drug Interaction between Omeprazole or Esomeprazole and Clopidogrel. Ph.D. Thesis, University of Kansas, Lawrence, KS, USA, 23 April 2015.
- Ogilvie, B.W. An In Vitro Investigation into the Mechanism of the Clinically Relevant Drug-Drug Interaction between Omeprazole or Esomeprazole and Clopidogrel. Ph.D. Thesis, University of Kansas, Lawrence, KS, USA, 23 April 2015.