The precise regulation of genetic information, as it is passed from gene to transcript to protein, is crucial for the survival of cells and organisms. From a single gene, multiple mature messenger RNA (mRNA) transcripts arise through alternative pre-mRNA, resulting in mature species with differences in both the coding and non-coding regions
[1]. Even beyond the end-points of mRNA transcription, the quality and quantity of mRNAs in cells is tightly controlled through various pathways
[2]. Nonsense-mediated mRNA decay (NMD) is a critical cellular surveillance mechanism that recognizes and eliminates aberrant RNAs containing premature termination codons (PTC) or abnormally long 3′ untranslated regions (UTRs). NMD was first found to affect one-third of the mutated mRNAs
[2]. Transcripts with destabilizing PTC in their coding region are products of endogenous genes with nonsense or frameshift mutations, pseudogenes
[3], or from alternative splicing events leading to intron retention or inclusion of PTC-containing exons
[4]. To avoid producing C-terminally truncated proteins that can have deleterious effects for the organism, those transcripts harbouring PTC are recognized and subsequently degraded
[5][6][5,6].
In mammalian cells, the discrimination of PTC-containing transcripts depends on the position of PTC in mRNA. Transcripts containing PTC at least 50–55 nucleotides upstream of the last exon-exon junction are recognized as “premature” and degraded through NMD. As a caveat, this definition changes across the species. In
Saccharomyces cerevisiae, PTC is defined independently of exon boundaries
[5]. In another variation, the presence of introns is not necessary to define PTCs in
Drosophila or in
Caenorhabditis elegans, which shows a mechanistic diversity in the initiation of the NMD pathway
[5].
NMD is a cytoplasmic and translation-dependent process. During pre-mRNA splicing, a multi-subunit protein complex, spanning ∼20–24 nucleotides, is deposited upstream of the exon-exon junction; the exon junction complex (EJC). Associated to mRNA, EJCs are transported into the cytoplasm, where the force of the ribosome, as it translates the transcript, is sufficient to remove the EJCs. Transcriptome-wide analysis and biological studies showed that EJCs are not loaded equally across all exon junctions of a transcript
[7]. During translation of a normal transcript, the stop codon in the last exon ensures that no EJCs remain on the mRNA upon translation termination. The position of the ribosome at the end of the transcript is also important for translation termination, where interactions to proteins bound to the mRNA poly(A) tail and release factors are required. A stalled ribosome at a PTC leaves remaining downstream EJCs
[2] and a distance to the 3′-end and poly(A) tail may be too large to facilitate termination. The resulting delayed release of the ribosome from the transcript affords the time needed to assemble NMD-related proteins and recruit other cofactors
[8].
1.1. The NMD Machinery
The NMD pathway was first elucidated using unbiased genetic screens from
Caenorhabditis elegans and
Saccharomyces cerevisiae [9][10][9,10]. Seven genes were identified in nematodes, termed
SMG1–7 (suppressor with morphological effect on genitalia proteins 1–7). Mutations to SMG were non-lethal, indicating that NMD is not essential in nematodes
[9]. Three orthologous genes to
SMG2, SMG3 and
SMG4,
UPF1–3 (up-frameshift 1–3), were identified in
S. cerevisiae [10]. Homology searches continued to identify orthologous genes in other species, including
Arabidopsis,
Drosophila and mammals
[11].
In humans, NMD members include the hUPFs—human up-frameshift (UPF) proteins (UPF1, UPF2, UPF3a and UPF3b), the suppressors with morphological effects on genitalia proteins (SMG1, SMG5, SMG6, SMG7, SMG8 and SMG9), and the exon junction complex (EIF4A3, MAGOH, RBM8A and Barentsz (BTZ)) (a)
[2][12][13][14][2,12,13,14]. The EJC complex recruits the evolutionarily conserved UPF proteins and plays an essential role in NMD
[15]. During the pioneer round of translation, some EJC components are displaced by the ribosome, and this positional information by EJC is preserved until the mRNA is translated
[15][16][15,16]. In the presence of a PTC, translation pauses upstream of an EJC and the eukaryotic release factors (eRF) physically bind and recruit UPF1 (the RNA helicase)
[17][18][19][17,18,19]. The eRFs recognize the stop codon, and when the mRNA stop codon enters the ribosomal A site, the termination of the protein synthesis occurs. The single eukaryotic class-I RF eRF1 recognizes all three (UAG, UGA, UAA) stop codons
[20].
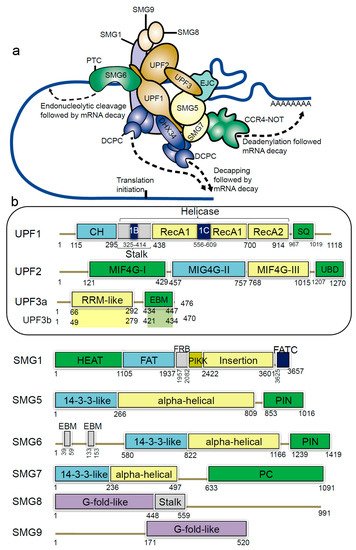
Figure 1. Schematic representation of domains and motifs of the nonsense-mediated mRNA decay (NMD) factors. (
a) The NMD complex UPF: up-frameshift; SMG: suppressor of morphogenetic effect on genitalia; DHX34: DEAH box polypeptide 34; DCPC: the decapping complex; EJC: exon junction complex; CCR4-NOT: carbon catabolite repressor protein 4 (CCR4)–NOT deadenylase complex
[21][31]. (
b) For the UPF and SMG proteins: CH: cysteine-histidine rich domain; Stalk: RecA1 domain by two long ‘stalk’ helices; RecA1 and RecA2: RecA-like domains; 1B and 1C: subdomains within the helicase core; SQ: serine-glutamine rich domain; RRM: RNA recognition motif; EBM: exon junction binding motif; MIF4G: middle of 4G-like domains; UBD: UPF1-binding domain; PIN: PilT N-terminus domain; PC: C-terminal proline-rich region; HEAT: Huntingtin, elongation factor 3 (EF3), protein phosphatase 2A (PP2A), yeast kinase TOR1 domain; FAT: focal adhesion kinase domain; FRB: FKBP12-rapamycin-binding; PIKK: phosphatidylinositol 3-kinase-related protein kinase domain; FATC: C-terminal FAT domain; G-fold-like: domains involved in dimerization between SMG8-SMG9
[21][22][23][31,32,33].
Initiation of the NMD pathway leads to remodelling of the surveillance complex (SURF), which includes the UPF1, SMG1, eRF1 and eRF3 proteins. UPF3b attaches to the EJC and anchors UPF2. The SURF complex binds with the UPF2, UPF3b and an EJC downstream of the PTC, forming the decay-inducing complex (DECID)
[24][21]. Along with the UPF proteins the SURF complex promotes the phosphorylation of UPF1 by SMG1. In contrast, for the dephosphorylation of UPF1 a multiprotein complex composed of SMG5, SMG6, SMG7 and protein phosphatase 2A is required
[25][22]. Allowing for the fine-tuning of the NMD activity, the UPF3a protein inhibits NMD, and this activity is regulated by the UPF3b protein
[26][23].
The main component of the NMD machinery is the UPF1/SMG2 protein, an ATP-dependent RNA helicase, which undergoes cycles of phosphorylation and dephosphorylation that are essential for NMD progression. The UPF1 protein is involved in the translation termination complex, when an EJC lies downstream of a termination event. UPF1 undergoes a large conformational change upon binding with UPF2 protein, which activates its RNA-helicase activity
[27][28][29][24,25,26]. Once the RNA-helicase is active, the RNA is exposed for degradation. The DEAH box polypeptide 34 (DHX34; a), an RNA helicase of the DEAH box family, associates with several components of the NMD complex in cell lysates, and preferentially binds with the hypophosphorylated UPF1
[30][31][32][27,28,29]. It is proposed that DHX34 is involved in the activation of UPF1 phosphorylation, and mediates a change in interaction patterns within the NMD, which propagates NMD activation
[31][32][33][28,29,30].
There are many pathways that lead to degradation of NMD-targeted RNAs. Studies show that in yeast, PTC-containing transcripts are degraded predominantly through deadenylation-independent process involving decapping by the Dcp1p/Dcp2p enzyme and 5′–3′ exonucleotic digestion by Xrn1p
[6][29][6,26]. In human cells, those transcripts are degraded through multiple mechanisms, such as endonucleolytic cleavage
[30][27], exosome mediated 3′–5′ decay
[33][30] or deadenylation-dependent decapping
[29][26]. Lykke-Andersen et al. performed a transcriptome-wide identification of NMD substrates and their 5′–3′ decay intermediates to establish that SMG6-catalyzed endonucleolysis widely initiates the degradation of human nonsense RNAs, whereas decapping is used to a lesser extent
[21][31].
1.2. Structural Insights of NMD Components at a Glance
The UPF1 protein has a conserved cysteine-histidine-rich domain (CH-domain), followed by two RecA-like domains (RecA1 and RecA2; helicase region), and a SQ (serine-glutamine) domain (b)
[23][34][33,34]. From the structural analysis it is known that binding of UPF2/UPF3 protein to the CH-domain of UPF1 activates UPF1 ATPase and the helicase activities
[35] (). The UPF2 structure consists of four core regions, three domains are the middle portion of eukaryotic initiation factor 4-gamma (MIF4G-1, 2 and 3) domains and a C-terminal domain. This C-terminal domain of the UPF2 protein plays an important functional role, as it binds to the UPF1 CH-domain, enhancing its helicase activity
[34]. Particularly, the MIF4G-3 domain interacts with the RRM (RNA recognition motif) domain of the UPF3b protein (b)
[36], as well as the SMG1 protein interacts with the MIF4G-3 domain at the same time as UPF3b, but in a non-competitive way
[37]. Both UPF3a and UPF3b proteins do not show direct binding to the RNA, despite having a RNP domain (ribonucleoprotein or RRM) at the N-terminus (b)
[38].
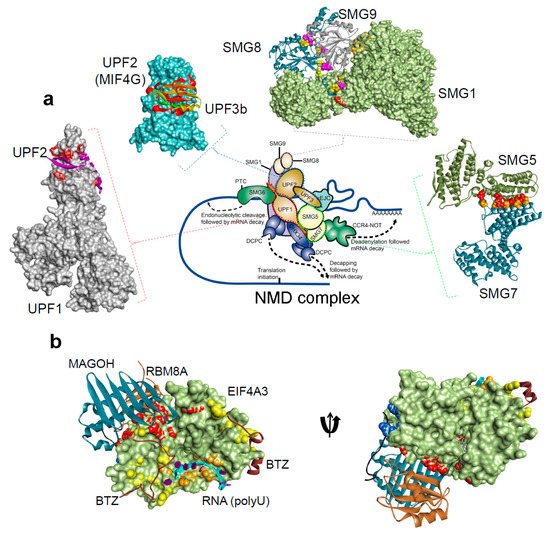
Figure 2. The protein-protein and protein-RNA binding interface for the NMD components, from the Protein Data Bank database (
http://www.rcsb.org/pdb)
[21][22][23][34][36][39][40][41][42][43][44][45][46][47][48][31,32,33,34,36,103,104,105,106,107,108,109,110,111,112]. (
a) NMD pathway schematic representations, and protein-protein interactions form the complex: UPF1-UPF2 (PDB: 2wjv)
[34], UPF2-UPF3b (PDB: 1uw4)
[36], SMG5-SMG7 (PDB: 3zhe)
[41][48][105,112], SMG8-SMG9 (PDB: 5nkk)
[42][106], SMG1–SMG8–SMG9 (PDB: 6syt)
[43][107]. (
b) The exon junction complex; Mago-Y14-eIF4AIII-Barentsz-UPF3b (PDB: 2xb2)
[44][108] (). The H-bond analysis was performed using BIOVIA Discovery Studio Visualizer program [Dassault Systemes, BIOVIA Corp., San Diego, CA, USA].
Table 1. The crystal structures available for the protein-protein binding interface available for different NMD components, from the Protein Data Bank database (
http://www.rcsb.org/pdb)
[47][111]. UPF: up-frameshift; SMG: suppressor of morphogenetic effect on genitalia.
| Protein Interacting Partners |
PDB ID. |
Resolution |
Method |
References |
| UPF1-UPF2 |
2wjv |
2.85 Å |
X-Ray diffraction |
[34] |
| UPF2-UPF3b |
1uw4 |
1.95 Å |
X-Ray diffraction |
[36] |
| SMG5-SMG7 |
3zhe |
3 Å |
X-Ray diffraction |
[48][112] |
| SMG8-SMG9 |
5nkk |
2.64 Å |
X-Ray diffraction |
[42][106] |
| SMG1–SMG8–SMG9 |
6syt |
3.45 Å |
Electron microscopy |
[43][107] |
| Mago-Y14-eIF4AIII-Barentsz-UPF3b |
2xb2 |
3.4 Å |
X-Ray diffraction |
[44][108] |
The UPF1 protein is phosphorylated by the phosphoinositide 3-kinase related kinase (SMG1)
[49][39]. SMG1 further associates with two cofactors, SMG8 and 9, and eukaryotic release factors eRF1 and 3a
[24][21]. As a result the phosphorylation of UPF1 recruits SMG5/6/7 proteins, and these recruited components share a phosphoserine-binding domain
[50][40]. The functional dependency between phosphorylation or dephosphorylation cycle and the ATPase or the helicase activities of the UPF1 protein, is an interesting area that needs investigation. The interactions between SMG5-SMG7 results in a stable heterodimer complex
[51][41]. Composing of two EJC-binding motifs (EBMs) the SMG6 protein harbours an endonuclease activity that cleaves the PTC-mRNA (b)
[52][42]. Identifying the structural and the functional relationship between UPF1, SMG5, SMG6, SMG7 and their interacting proteins would be an interesting area to investigate
[53][43].
1.3. NMD Target Selection, more than just Coding Transcripts
Despite an increased understanding of the NMD process, questions remain around the rules governing the NMD target selection. Varying between the organism or cell type, ~5–20% of the transcripts can be subjected to NMD and these targets extend beyond the classical understanding of this machinery. Beyond mRNA with PTC in coding regions, a number of features can target different RNA-species for degradation by NMD. Additional targets can be classified into three main categories: (1) transcripts with destabilizing PTC arising in pseudogenes
[5], or from alternative splicing events leading to the intron retention or inclusion of PTC-containing exons
[4]. (2) Transcripts with limited or no clear coding potential, such as small RNAs derived from intragenic regions
[54][44], long non-coding RNAs
[21][31] and mRNAs of inactivated transposons
[5]. (3) Transcripts with upstream open reading frames, or with abnormally long 3′UTRs, or wild-type mRNAs with no atypical features
[5]. It has been demonstrated that the NMD process can occur even without the presence of EJC bound to the mRNA. In this pathway, UPF1 binds to the 3′ UTRs of the transcript and interacts with exon–exon junction components found in the cytoplasm or EJC stably associated with 3′ UTRs. The mechanism of this process is still not fully understood.
Lykke-Andersen et al. performed a transcriptome-wide identification of NMD substrates in HEK293 cell line and found that genes hosting small nucleolar RNAs (snoRNAs) and microRNAs (miRNAs) were significantly enriched among NMD substrates. The researchers hypothesized that snoRNA host genes need to be highly transcribed to regulate the high demand for snoRNA production and that the expression of individual snoRNAs and their cognate spliced RNA can be uncoupled through alternative splicing and NMD
[55][45].
Studies of long non-coding RNAs indicate that many contain long regions located downstream of the stop codon that are unprotected by ribosomes. Considering that a long 3′ UTR triggers NMD, translation termination upstream of these ribosome-free regions gives a mechanistic explanation for the recognition and elimination of non-coding RNAs by the NMD pathway
[56][46].