It has been long recognized that cancer cells reprogram their metabolism under hypoxia conditions due to a shift from oxidative phosphorylation (OXPHOS) to glycolysis in order to meet elevated requirements in energy and nutrients for proliferation, migration, and survival. However, data accumulated over recent years has increasingly provided evidence that cancer cells can revert from glycolysis to OXPHOS and maintain both reprogrammed and oxidative metabolism, even in the same tumor. This phenomenon, denoted as cancer cell metabolic plasticity or hybrid metabolism, depends on a tumor micro-environment that is highly heterogeneous and influenced by an intensity of vasculature and blood flow, oxygen concentration, and nutrient and energy supply, and requires regulatory interplay between multiple oncogenes, transcription factors, growth factors, and reactive oxygen species (ROS), among others. Hypoxia-inducible factor-1 (HIF-1) and AMP-activated protein kinase (AMPK) are key modulators of the switch between reprogrammed and oxidative metabolism. Our review focuses on cross-talks between HIF-1, glucose transporters (GLUTs), and AMPK, and other regulatory proteins including oncogenes such as c-Myc, p53, and KRAS along with growth factor-initiated protein kinase B (PKB)/Akt, phosphatidyl-3-kinase (PI3K), and mTOR signaling pathways in controlling cancer cell metabolism.
Note: The following contents are extracted from your paper. The entry will be online only after author check and submit it.
1. Introduction
Cancer cells often suffer from hypoxia, nutrient (glucose and amino acid), and energy deprivation resulting from insufficient vasculature and blood supply
[1]. These stress conditions are key factors imposed on proliferating tumor cells to trigger their malignant transformation and to enable them to overcome or escape antitumor immune surveillance and to avoid cellular senescence and apoptosis
[2][3][4][2,3,4]. This results in tumor progression and aggressiveness, genetic instability, development of chemo- and radio-resistance, and poor prognosis
[5][6][5,6].
Under physiological conditions, oxidative phosphorylation (OXPHOS), that is, coupling of oxidation reactions with mitochondrial electron transportation chain (ETC), is the most efficient source of ATP production, generating a much larger amount of energy as compared to anaerobic glycolysis; however, under hypoxic conditions, glycolysis is the only process that provides cells with energy
[7][8][7,8]. In the hypoxic microenvironment, cancer growth is maintained by metabolic and bioenergetic reprogramming that is characterized by an adaptive switch from OXPHOS to glycolysis followed by excessive glucose consumption and lactate production. This phenomenon was first discovered by German scientist Otto Warburg in 1927 and was denoted as the Warburg effect by Efraim Racker in 1972
[9][10][11][9,10,11].
Molecular mechanisms underlying cancer cell tolerance to prolonged hypoxia and nutrient/energy starvation are very complex and can work both at transcriptional and post-translational levels. Hypoxia-inducible factor-1 (HIF-1) is a master regulator of cellular oxygen sensing and adaptation to hypoxia and a ubiquitous transcriptional activator that regulates the expression of numerous genes at DNA and epigenetic (chromatin remodeling/histone modification) levels
[12][13][14][12,13,14]. The modulation of gene expression by HIF-1 causes alterations in mitochondrial oxidative metabolism, glucose uptake and oxidation, energy production, and angiogenesis in order to enable cancer cell proliferation, migration, and survival.
However, a large body of data has shown that most tumors grow in their interaction with a highly heterogeneous microenvironment with different densities of blood and lymph vessels, amount and types of infiltrating cells, extracellular matrix composition, and content of signaling molecules, among others
[15] Moreover, many tumors are not monoclonal despite originating from a single cell; instead, they are composed of multiple distinct clones that vary by morphological and phenotypic features depending on cancer type, cancer stage, and treatment regimes, among other factors
[16][17][18][16,17,18]. This phenomenon denoted as tumor heterogeneity implies that a heterogeneous population of various cell types with distinct gene expression and metabolic profiles as well as proliferative, angiogenic, and metastatic potential co-exist within a definite tumor.
Furthermore, results of experimental, bioinformatics, and computer/mathematical modeling approach increasingly evidence that cancer cells do not fully rely on glycolysis; instead, they preserve oxidative metabolism
[19][20][19,20]. This indicates that cancer cells acquire hybrid or heterogeneous metabolism, which enables them to use both glycolysis and OXPHOS as sources of ATP and that oxidative catabolic pathways including tricarboxylic acid (TCA) cycle (Krebs cycle), oxidative decarboxylation of pyruvate, glutaminolysis, and fatty acid β-oxidation (FAO) can remain functional as sources of reducing equivalents (NADH and FADH
2), as well as carbon and nitrogen atoms
[20]. Moreover, multiple switches between the metabolic pathways can exist depending on various intrinsic and extrinsic factors such as nutrient and energy availability, micro-environmental and dietary factors, and clinicopathological characteristics such as tumor stage, histological type, differentiation grade, lymph node involvement, and depth of invasion, among others. Importantly, these metabolic switches impact tumor outcome and patient responsiveness to anti-cancer therapy.
To provide the cancer cell metabolic plasticity, induction of numerous genes and activation or inhibition of multiple oncogenes, growth factors, and tumor suppressors are required
[21]. A crucial role in this phenomenon belongs to the interplay between HIF-1 and AMP-activated protein kinase (AMPK), an energy sensor and master regulator of cellular metabolism and bioenergetics. AMPK is a heterotrimeric serine/threonine kinase that is activated in response to decreasing in AMP/ATP ratio in order to provide ATP production through both glycolysis and OXPHOS
[22]. In general, AMPK maintains ATP level in cells due to a switch from anabolic to catabolic metabolism through the stimulation of glucose uptake, rate of aerobic glycolysis, and mitochondrial oxidative metabolism via β-oxidation of fatty acids
[23].
Additionally, both hypoxia and nutrient deprivation can cause elevated generation of reactive oxygen species (ROS) by mitochondrial ETC and Nox family NADPH oxidases, resulting in oxidative stress and alterations in cell signaling pathways
[24]. A variety of ROS types can affect the activities of both HIF-1 and AMPK along with intracellular effectors of cell signaling pathways and transcription factors to trigger cancer progression and metastasis under hypoxia, nutrient/energy starvation, and oxidative stress conditions
[25].
2. Hypoxia-Inducible Factors
The master regulators of oxygen homeostasis, hypoxia-inducible factors (HIFs), are evolutionarily conserved transcription factors that are expressed in all eukaryotes in three isoforms: HIF-1α, HIF-2α, and HIF-3α (reviewed by
[26][27][26,27]). HIF-1α undergoes heterodimerization with HIF-1β, both containing basic helix-loop-helix (bHLH) domains along with Per-aryl hydrocarbon nuclear translocation (ARNT)-Sim homology (PAS) domain
[28]. Under normoxic conditions, HIF-1α is destabilized through continuous degradation by ubiquitin-proteasome system (UPS)
[29]. However, under acute hypoxia conditions, HIF-1α dimerizes with HIF-1β and becomes stabilized in order to undergo translocation into the nucleus and to bind to hypoxia response elements (HREs) on DNA for regulation of gene expression. This leads to the over-expression of key regulatory enzymes of glycolysis and pentose phosphate pathway (PPP), as well as to down-regulation or mutations in genes encoding pyruvate decarboxylase complex (PDC) and TCA cycle enzymes or ETC enzymatic complex I, as observed in various cancer types (reviewed by
[30][31][32][30,31,32]).
Under normoxic conditions, two proline residues of HIFα subunits undergo hydroxylation by prolyl-hydroxylase domain proteins (PHDs) that exist in three isoforms, from PHD1 to PHD3, with distinct functions
[33]. Under chronic hypoxia conditions, over-expression and hyper-activation of PHDs are followed by HIFα desensitization and cell protection from necrosis
[34]. Hydroxylation of proline residues and acetylation of lysine residue enables a recognition of HIF1α, HIF2α, and HIF3α by von Hippel–Lindau protein (pVHL) and the recruitment of E3 ubiquitin ligase for the HIFα polyubiquitination and UPS-mediated degradation
[35]. Genetic loss in the pVHL tumor suppressor has been shown to cause HIF-1 stabilization and activation, even under normoxia conditions followed by tumor cell proliferation and survival
[36][37][36,37]. Additionally, hydroxylation of asparagine residue by the factor inhibiting HIF (FIH) prevents binding of coactivator p300/CBP, followed by a decrease in HIFα transcriptional activity
[38].
With the use of the tumor metabolism modeling approach, it has been shown that in the hypoxic microenvironment, both intracellular and environmental factors contribute to metabolic reprogramming of cancer cells and that various growth factor-initiated cell signaling cascades and transcription factors can affect HIF-1 activity
[39]. An interplay between HIF-1 and a variety of oncogenes such as Ras, c-Myc, p53, AMPK, along with PKB/Akt, PI3K, and mTOR signaling pathways has been observed to control mitochondrial ETC functioning and energy production to maintain cancer cell proliferation and survival
[40][41][42][43][44][45][46][47][40,41,42,43,44,45,46,47].
For example, an interplay between HIF-1α and p53, two transcription factors regulated by both E3 ubiquitin ligase and murine double minute 2 (Mdm2), in response to hypoxia during carcinogenesis has been shown
[48]. The p53 activation by gamma-rays used in cancer treatment triggers Mdm2-mediated HIF-1α UPS-mediated degradation. This leads to a decrease in the peroxisome proliferator-activated receptor-gamma co-activator 1β (PGC-1β) inhibition and promotes mitochondrial biogenesis
[49]. Additionally, oncogenic KRAS induces HIF-1α and HIF-2α genes, which leads to decreased OXPHOS and ATP production and increased mitochondrial ROS generation in colon cancer cells
[50]. Moreover, KRAS can enhance ROS generation by NADPH oxidases, for example, in a Rac1-Nox4-dependent manner
[51].
Under hypoxic conditions, HIF-1α expression can associate with the growth factor expression. Indeed, signal transduction pathways initiated by binding of epidermal growth factor (EGF) and platelet-derived growth factor (PDGF) to their membrane-bound receptors, receptor tyrosine kinases (RTKs), activate Ras/PI3K/phosphatase and tensin homolog (PTEN)/Akt and mTOR signaling to induce HIF-1 and c-Myc expression
[52][53][52,53]. For example, PI3K/Akt/mTOR pathway-mediated stimulation of HIF-1 mRNA translation through the activation of two downstream targets of mTOR, p70SK6, and 4E-BP1 in breast cancer cells has been observed
[37].
Additionally, Krüppel-like factor 5 (KLP5) promotes non-small cell lung cancer (NSCLC) cell apoptosis via direct suppression of HIF-1α and glycolysis. The over-expression of KLF5 promotes cancer cell survival and hypoxia-induced cisplatin resistance through the activation of the PI3K/Akt/mTOR pathway
[54]. Further, the mechanism of PDC inhibition and reduction during the oxidative decarboxylation of pyruvate to acetyl-CoA in response to hypoxia may involve the mitochondrial PKB/Akt accumulation and pyruvate dehydrogenase kinase 1 (PDK1) phosphorylation at Thr-346. This triggers glycolysis to maintain tumor cell proliferation, to inhibit apoptosis and autophagy
[55].
Moreover, in tumor patients, HIF-1α expression correlates with tumor stage and vascularization, lymphatic invasion, metastasis, and vascular endothelial growth factor (VEGF) expression as shown in many cancer types such as esophageal squamous cell cancer, as well as colorectal and hepatocellular carcinomas
[56][57][58][59][56,57,58,59]. Moreover, the inhibition of HIF-1α and VEGF expression both at mRNA and protein levels suppress tumor growth, therefore targeting mTOR/HIF-1α/VEGF is a promising strategy in anti-cancer therapy
[58]. Indeed, the inhibition of human cervical cancer growth and the enhancement of tumor radio-sensitivity can be achieved by down-regulation of HIF-1α and VEGF and up-regulation of p53
[60]. In an animal model of cervical cancer, the tumor growth inhibition by formononetin and cisplatin associates with the decrease in HIF-1α and VEGF expression
[61]. Targeting HIF-2 in clear cell renal carcinoma (ccRCC) led to dissociation of HIF-2α/HIF-1β dimer to suppress tumor growth
[62].
Further, metabolomics and quantitative proteomics approaches have shown that the mitochondrial NAD
+-dependent deacetylase family of enzymes, sirtuins (SIRTs), can alter cellular metabolism and revert the Warburg effect in tumor cells
[63]. For example, SIRT3 over-expression associates with kidney cancer growth inhibition and maintaining mitochondrial homeostasis, as well as modulating ROS production to sensitize biomolecules and cells to oxidative damage
[64]. Whereas low SIRT3 level associates with poor differentiation and unfavorable prognosis in primary hepatocellular carcinoma (HCC)
[65]. Moreover, anti-cancer agent etoposide-induced genotoxic-mediated cancer cell apoptosis correlates with both SIRT1 and SIRT3 over-expression
[66].
SIRTs can regulate mitochondrial metabolism in response to diverse nutrient signals through the deacetylation of various proteins including TCA cycle enzyme isocitrate dehydrogenase-2 (IDH2), ETC enzymatic complexes, and antioxidant enzyme Mn-superoxide dismutase (SOD2). Thus, they promote the TCA cycle and ETC functioning, and reduce ROS generation, while a loss of SIRTs enhances metabolic reprogramming in cancer cells through destabilization of HIF-1α to downregulate glycolytic genes
[67]. For example, in breast cancer, the over-expression of SIRT3 decreases the rate of glycolysis and inhibited cell proliferation followed by tumor suppression, whereas down-regulation of SIRT3 increases ROS production leading to HIF-1α stabilization and upregulation of HIF-1α target genes
[68]. Seemingly, SIRT3 can regulate metabolic reprogramming in cancer cells also through deacetylation of p53 transcription factor to promote its UPS-mediated degradation, as shown in phosphatase and tensin homolog (PTEN)-deficient non-small cell lung cancer cells
[69].
3. An interplay between HIF-1 and Facilitative Glucose Transporters
The phenotypic hallmark of more than 90% of primary and metastatic tumors is an increase in glucose uptake from the blood, which occurs to compensate for a low ATP yield via glycolysis and to a great extent depends on facilitative glucose transporters (GLUTs) encoded by the
SLC2A family of genes
[70]. This family comprises 14 members, GLUT1–14, grouped into four classes on the basis of sequence similarity. Additionally, GLUTs vary in their affinity to glucose, regulation, tissue distribution, and expression level under both physiological and pathological conditions.
Under physiological conditions, GLUT4 is a major insulin-sensitive glucose transporter. TBC1D1, Tre2/Bub2/Cdc15 (TBC) domain family member 1 protein, can regulate insulin-stimulated GLUT4 translocation into a mammalian cell membrane, thereby triggering glucose uptake
[71]. TBC1D1 is a Rab-GTPase-activating protein and contains
N-terminal phosphotyrosine-binding (PTB) domains and a
C-terminal Rab-GTPase (GAP) domain. In response to insulin, TBC1D1 is phosphorylated by Akt and AMPK, however, this does not alter intrinsic Rab–GAP activity
[72].
The efficacy of glucose uptake by cancer cells depends mainly on the activities of GLUT1 and GLUT3 and, to a lesser extent, GLUT4 and GLUT10
[73]. For example, higher expression of GLUT1 and GLUT3 in papillary carcinoma, as compared to follicular carcinoma and non-neoplastic thyroid lesions, has been reported
[74]. Additionally, both GLUT1 and GLUT3 have been up-regulated in poorly differentiated endometrial and breast cancers, both at mRNA and protein levels
[75]. Transactivation of GLUT3 occurs in a Yes-associated protein (YAP)-dependent manner, suggesting that this pathway serves as a regulator of metabolic reprogramming during cancer progression, and, thus, can be considered as a promising anti-cancer therapeutic target
[76].
GLUT1 localization in a cell membrane can be increased by phosphorylation of thioredoxin-interacting protein (TXNIP), an α-arrestin family member. The elevated glucose concentration induces expression of TXNIP, which inhibits glucose uptake directly by binding to GLUT1 and stimulating its endocytosis via clathrin-coated pits or indirectly by reducing GLUT1 biosynthesis at the mRNA level. AMPK has been shown to cause phosphorylation and rapid degradation of TXNIP, thereby increasing GLUT1 function
[77]. In addition to gene expression regulation at the DNA level, histone modifications can contribute to the modulation of glucose transporter induction. For example, epigenetic regulation of the expression of the
SLC2A1 gene encoding GLUT1 can be due to the induction of the
HDAC2 gene by beta-hydroxybutyrate, a ketone body, to enhance H3K9 acetylation under starvation conditions in brain tissue
[78].
GLUT3 induction during epithelial-to-mesenchymal transition (EMT) by ZEB1 transcription factor promotes NSCLC cell proliferation
[79]. Additionally, in NSCLC cell culture and in an in vivo model, increased glucose uptake with the involvement of GLUT3 and caveolin 1 (Cav1), an important component of lipid rafts, triggers tumor progression and metastasis. Interestingly, Cav1-GLUT3 signaling can be targeted by atorvastatin, an FDA-approved statin, which decreases cholesterol biosynthesis due to the inhibition of 3-hydroxy-3-methyl-glutaryl-CoA reductase, and this reduces EGFR-tyrosine kinase inhibitor (TKI)-resistant tumor growth and increases the overall patient survival
[80].
The expression level of GLUT1 correlates with that of HIF-1α in many cancer types, including colorectal and ovarian cancers, and this associates with tumor clinicopathological characteristics such as tumor size, location, and patient age and gender; however, there can be differences in the intracellular location of these two proteins
[81][82][81,82]. For example, GLUT1 was found in membranes of multifocally necrotizing cancer cells and in the cytoplasm of cancer cells with no necrosis, whereas HIF-1α mostly had a cytoplasmic location
[82]. Immunoreactivity of GLUT1 was significantly higher in node-positive colorectal cancer compared to node-negative one.
Additionally, an interplay between GLUTs, HIF-1, and glycolytic enzymes has been observed in many cancer types. For example, HIF-1α expression has been reported to correlate positively with those of both GLUT1 and lactate dehydrogenase-A (LDH-A or LDH-5) at both mRNA and protein levels in human gastric and ovarian cancers, and this was found to be associated with tumor size, depth of invasion, distant metastasis, clinical stage, and differentiation status
[83][84][83,84]. Additionally, a correlation between the expressions of GLUT1, VEGF, and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases-3 and -4 (PFKFB-3 and PFKFB-4) has been observed in gastric and pancreatic cancers. GLUT3 induction also correlates with the over-expression of glycolytic enzymes including hexokinase 2 (HK2) and pyruvate kinase M2 (PKM2), which are associated with cancer invasiveness, metastasis, and poor prognosis
[85].
4. Role of HIF-1 in Metabolic Reprogramming of Cancer Cells
4.1. Enhancement of Glycolysis
As early as 1925, C. Cori and G. Cori found glucose content was 23 mg less and the content of lactate was 16 mg greater than those in veins of normal tissues when studying the axillary veins of hens with Rous sarcoma
[86]. Afterward, Otto Warburg and co-workers compared glucose and lactate concentrations in tumor veins and arteries and found 69 mg greater lactate in the vein blood than that in the same volume of aorta blood of rats with Jensen sarcoma, whereas glucose uptake by the tumor tissue was 52–70% and by normal tissues was 2–18%
[9].
The Warburg effect has been experimentally confirmed by the overexpression of glycolytic enzymes accompanied by a deficit in OXPHOS-mediated ATP production in many cancer types in both cultured cell lines and animal models
[87][88][87,88]. Genes affected by HIF-1 and implicated in carcinogenesis include the
SCL2A solute carrier family and those encoding glycolytic enzymes such as HK2, phosphofructokinase 1 (PFK1), fructose-bisphosphate aldolase A (ALDOA), α-enolase (ENO1), PKM2, and LDH-A as well as genes encoding PDK and enzymes of pentose phosphate pathway (PPP)
[89][90][89,90].
The first reaction of glycolysis () is catalyzed by the key rate-limiting enzyme, hexokinase, which has four isoforms in mammalian cells, among which HK2 is overexpressed at both mRNA and protein levels in many tumor types including HCC, ovarian cancer, and others
[91][92][93][91,92,93]. Furthermore, correlation of overexpression and co-localization of both HK2 and HIF-1α in cancer cells near necrosis regions have been reported.
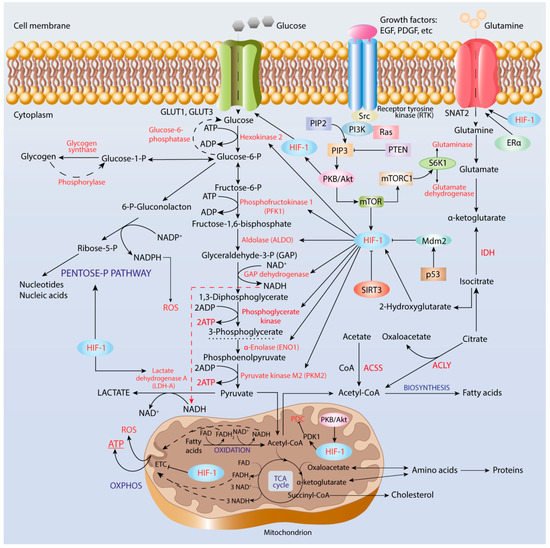
Figure 1. Regulation of metabolic reprogramming in cancer cells. Hypoxia-inducible factor-1 (HIF-1) induces expression of genes, which encode glucose transporters, GLUT1 and GLUT3, enzymes of glycolysis and pentose-phosphate pathway, and pyruvate dehydrogenase complex kinase. The activity of HIF-1 is regulated by the Ras-protein kinase B (PKB)/Akt-mammalian target of the rapamycin (mTOR) axis.
The second key rate-limiting glycolytic enzyme is PFK, a tetrameric enzyme in mammals that catalyzes the third reaction of glycolysis, that is, phosphorylation of fructose-6-phosphate to fructose-1,6-bisphosphate (FBP) accompanied by ATP utilization. The interplay between HIF-1α and Ras/Src oncogenes in the tumor microenvironment in the regulation of PFK1 and PFK2 isoenzymes has been suggested as being a contributor to human cancer cell proliferation and survival
[94]. PFK1 is an allosteric enzyme activated by fructose-2,6-bisphosphate that is produced from fructose-6-phosphate by the bifunctional phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB) family of enzymes that are induced by HIF-1α. PFKFB3 has the highest kinase activity, whereas PFKFB4 has more FBPase-2 activity and co-expression of PFKFB3 and PFKFB4 provides sufficient glucose metabolism. Thus, the PFKFB family enzymes are promising targets in anti-cancer therapy in order to combat tumor growth, invasion, and metastasis. For example, silencing the
PFKFB2 gene has been shown to significantly inhibit ovarian and breast cancer growth and to enhance paclitaxel sensitivity and patient survival
[95].
In the glycolytic pathway, there are two enzymes that catalyze the transfer of the phosphoryl group from a substrate to ADP, thereby producing ATP in the reaction of substrate-level phosphorylation, serving as an energy source in hypoxia conditions. The first enzyme is phosphoglycerate kinase (PGK), which catalyzes the conversion of 1,3-diphosphoglycerate to 3-phosphoglycerate. Several single nucleotide polymorphism variants of PGK1 with decreased catalytic efficiency and thermodynamic stability due to the alterations in local protein conformation have been found in carcinoma cells
[96]. The second enzyme is pyruvate kinase, which catalyzes the last reaction of glycolysis under aerobic conditions, that is, conversion of phosphoenol-pyruvate (PEP) into pyruvate, being allosterically regulated by fructose-2,6-bisphosphate. Four mammalian PK isoforms differing by regulation and tissue specificity, designated as PKM1, PKM2, PKR, and PKL, have been described
[97]. Among them, PKM2 is expressed in embryonic, proliferating, and tumor cells, and has a role in the progression of many cancer types such as ovarian, gastric, and lung cancers, among others.
[98][99][98,99]. PKM2 up-regulation has been shown to occur through mTOR-mediated HIF-1α stabilization and c-Myc-heterogeneous nuclear ribonucleoprotein (hnRNP)-dependent regulation
[100].
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) catalyzes the sixth reaction of glycolysis, that is oxidation of GAP to 1,3-biphosphoglycerate accompanied by reduction of NAD
+ to NADH. Regulation of GAPDH expression in cancer cells is not obvious. For example, GAPDH regulation has not been found in Hep-1-6 mouse hepatoma, Hep-3-B and HepG2 human hepatocellular carcinoma, A-549 human adenocarcinoma, and HT-29 and HCT-116 colon cancer cell lines
[101]. This indicates that GAPDH is not an attractive target in anti-cancer therapy and emphasizes the importance of the proper choice of housekeeping genes for the correct interpretation of experimental results.
Indeed, analysis and interpretation of transcriptomic data with the use of Cytoscape network showed highly over-expressed glycolytic genes including HK2, PFKP, ENO2, SLC2A3, SLC16A1, and PDK1 in patients with clear cell renal carcinoma
[102]. However, according to this study, some other glycolytic enzymes such as ALDOB, PKLR, PFKFB2, G6PC, PCK1, FBP1, and SUCLG1 were highly down-regulated. Therefore, a combination of several approaches including genomics, proteomics, metabolomics, and bioinformatics approaches should be used for the explanation of altered metabolic phenotype, bioenergetic signature, and increased glucose uptake resulting from both the activation of anaerobic glycolysis for cell proliferation and the impairment in mitochondrial functions of cancer cells
[103].
4.2. Pentose Phosphate Pathway
Other metabolic pathways can contribute to the Warburg effect by producing intermediates that fuel glycolysis. The overexpression of the PPP enzymes associated with HIF-1α stabilization and tumor progression has been reported to serve as an indicator of poor prognosis in cancer. Because PPP is linked to glycolysis, inhibition of PPP enzymes can serve as a promising strategy in anti-cancer therapy. For example, the ability of natural peptide carnosine to decrease the activities of PPP enzymes, as well as malate-aspartate and glycerol-3-phosphate shuttle mechanisms that carry electrons from glycolysis to ETC, has been observed in glioblastoma cell lines
[104].
PPP is a process that takes place in the cytoplasm and contains two branches: (i) oxidative branch yielding ribose-5-phosphate used in nucleotide and nucleic acid biosynthesis, giving rise to NADPH utilized in fatty acid biosynthesis, and (ii) non-oxidative branch giving rise to glyceraldehyde-3-phosphate (GAP) and fructose-6-phosphate, both of which can enter glycolysis
[31]. The first PPP reaction is the oxidation of glucose-6-phosphate to 6-phosphoglucono-δ-lactone catalyzed by a key time-limiting NADP-dependent enzyme, glucose-6-phosphate dehydrogenase (G6PDH). Up-regulation of G6PDH has been found in many cancer types and has been considered as a promising target for anti-cancer therapy to overcome cancer cell chemoresistance
[105][106][105,106]. For example, in human clear renal cell carcinoma, elevated glucose uptake and consumption, along with the increased activity of G6PDH and concentration of PPP-derived metabolites including NADPH, have been observed
[107].
The second oxidation reaction is the conversion of phosphoglucono-δ-lactone into ribulose-5-phosphate catalyzed by 6-phosphoglucono-δ-lactone dehydrogenase (6PGD), which is also over-expressed in many cancer types including lung and ovarian cancers
[108][109][108,109]. Up-regulated enzymes of the non-oxidative branch of PPP, such as thiamine pyrophosphate (TPP)-dependent transketolase family enzymes TKTL, TKTL1 and TKTL2, have also been found in various cancer types, including breast, lung, gastric, endometrial, head, and neck cancer
[110][111][112][113][114][110,111,112,113,114].
In addition to PPP, there are two NADP-dependent enzymes that produce NADPH: (i) isocitrate dehydrogenase (IDH) and (ii) decarboxylating malate dehydrogenase (malic enzyme). Both enzymes are associated with the TCA cycle and tumor growth. Over-expression of both ME1 and ME2 isomers of the malic enzyme has been found to cause a reduction in tumor suppressor p53 levels, however, down-regulation of ME2 caused a more prominent increase in ROS generation and phosphorylation/activation of p53 by AMPK followed by senescence, as compared to ME1
[115]. Importantly, NADPH is an essential component of NADPH oxidases, which represent a major source of ROS and produce superoxide anion radical (O
2•
−) as a primary product
[116].
4.3. Cancer Acidification and Its Role in the Reverse to OXPHOS
Typically, cells that are grown under in vitro culture conditions experience oxygen concentrations of 20%, the condition denoted as normoxia
[117]. However, under physiological conditions, the oxygen concentration in peripheral tissues can vary from 3.0% to 7.4%, with an average value of about 5.0% (38 mm Hg), the condition denoted as physioxia
[118]. Physiological oxygen concentration can transiently decrease under in vivo conditions due to various processes such as vasodilation and an increase in blood flow
[119]. Moreover, oxygen consumption rate can considerably vary between cell types depending on mitochondrial content and metabolic activity
[117].
Under physioxia conditions, pyruvate produced during glycolysis undergoes oxidative decarboxylation by pyruvate dehydrogenase complex (PDC) to yield acetyl-CoA and NADH. Oxygen deficiency condition (oxygen concentration less than 2%) decreases the efficacy of mitochondrial ETC to cause an increase in NADH/NAD
+ ratio, which triggers the conversion of pyruvate into lactate instead of its further oxidation by PDC. The over-production of lactate is due to the activity of the LDH isoforms (LDH-1 to LDH-5) differentially expressed in normal tissues and up-regulated during carcinogenesis
[120]. For example, increased glucose uptake, induction of glycolysis-related genes, excessive lactate production, and HIF-1α activation associated with aggressive phenotype and poor prognosis have been observed in patients with HCC and in Ewing sarcoma cells
[121][122][121,122]. These observations have led to the conclusion that forcing cancer cells into mitochondrial oxidative metabolism can efficiently suppress tumor progression, whereas targeting glycolytic enzymes can be an effective strategy to combat cancer growth.
Tissue lactate accumulation, up to 30–40 mM, leads to acidosis (pH ≤ 6.8), which is a hallmark phenotypic feature of tumor microenvironment affecting tumor progression, invasion, and metastasis
[123]. Normal cells cannot grow in an acidic microenvironment, however, acidosis is a necessary condition in order to promote cancer cell migration and invasion
[124][125][124,125]. Both endogenous and exogenous lactate is essential for the activation of certain enzymes such as matrix metalloproteinases, as well as for regulation of the expression of oncogenes (Myc, Ras), transcription factors (HIF-1, E2F1), tumor suppressors (BRCA1, BRCA2), and cell cycle genes, as shown in MCF-7 breast cancer
[126].
Tumor-derived lactate can inhibit activities of immune cells contributing to evasion of tumor cells of immune surveillance
[127]. Elevated LDH-A level is a negative prognostic cancer biomarker and is implicated in the enhancement of immune suppressive cells such as myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and dendritic cells (DCs)
[128]. However, lactate inhibits the activities of cytotoxic T-lymphocytes (CTLs) and natural killer (NK) cells. For example, the production of lactate by pancreatic cancer cells exerts immunosuppressive action due to inhibition of innate immune response via repressing cytotoxic activity of NK cells
[129].
In addition to lactate, carbon dioxide produced in catabolic pathways such as PPP contributes to the acidification of the tumor microenvironment. For example, in hypoxia conditions, tumor cells have been shown to produce more HIF-1-induced IX and XII isoforms of carbonic anhydrase, which catalyzes the reversible hydration of carbon dioxide into bicarbonate and protons to contribute to intracellular acidification and tumor cell survival
[130]. Moreover, in a mouse model of ductal carcinoma, in situ differences in levels of GLUT1 and carbonic anhydrase IX expression between normal and pre-cancer cells along with heterogeneity in intracellular pH values have been demonstrated
[131].
However, a growing body of data evidence that tumor cells demonstrate increased proton export due to the up-regulation of proton transporters such as Na
+/H
+ exchanger 1 (NHE1), H
+-lactate co-transporter, and monocarboxylate transporters (MCTs) to regulate intracellular pH values
[132][133][132,133]. For example, MCT1 serves as a prominent pathway for lactate uptake by human cervical squamous cell carcinoma, mouse model of lung carcinoma, and xenotransplanted human colorectal adenocarcinoma cells
[134]. Activities of these proton exchange systems represent additional adaptation and selection mechanisms, which enable emerging of chemoresistant cell clones and tumor progression and metastasis. Moreover, targeting these proton exchange molecules has potential in anti-tumor therapeutic strategies.
Importantly, due to the activities of lactate shuttle mechanisms, lactate can serve as energy fuel, important gluconeogenic substrate, and signaling molecule
[135]. For example, lactate can fuel the TCA cycle, as shown in human non-small cell lung cancers
[136]. In genetically engineered fasted mice lung and pancreatic cancer cells, the contribution of circulated lactate to the production of TCA cycle intermediates exceeded that of glucose, however, glutamine contributed more greatly than lactate in pancreatic cancer
[137].
Quantification of ATP amount produced via glycolysis and OXPHOS in nine randomly selected cancer cell lines demonstrated that in the lactic acidosis microenvironment (20 mM lactate, pH 6.7) ATP was generated almost twice as much by OXPHOS and almost four times less by glycolysis than that without lactic acidosis
[138]. Moreover, glucose consumption was much greater in the lactic acidosis environment than that without lactic acidosis in the same tumor cell lines. Therefore, lactate accumulation can serve as a mechanism that underlies the reverse from glycolysis to OXPHOS to produce ATP in cancer cells.
4.4. Lipid Biosynthesis
Under hypoxic conditions, the up-regulation of enzymes involved in fatty acid and cholesterol biosynthesis including citrate synthase, fatty acid synthase (FASN), and 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) is observed in many cancer types including pancreatic ductal adenocarcinoma (PDAC) and lung adenocarcinoma
[139][140][141][139,140,141]. A common single carbon source for both fatty acid and cholesterol biosynthesis is acetyl residue activated and carried by co-enzyme A, acetyl-CoA, which is supplied under physioxia condition by PDC. However, under hypoxia condition, there are two different ATP-dependent reactions that produce acetyl-CoA to link glucose and fatty acid metabolism: (i) cleavage of citrate by ATP citrate lyase (ACLY) to give rise to oxaloacetate and acetyl-CoA, and (ii) ligation of acetate by co-enzyme A catalyzed by acetyl-CoA synthase (ACSS)
[142][143][142,143].
ACLY is a key enzyme in cellular lipogenesis aberrantly expressed in many cancer types such as breast, liver, colon, lung, and prostate cancers. ACLY expression negatively correlates with tumor stage and differentiation, and this makes ACLY a potent target for anti-cancer therapy
[144]. ACLY links cellular metabolism to histone acetylation and, thereby, plays an important role in epigenetic regulation of cell functions
[145]. Under hypoxia conditions, histone acetylation and chromatin accessibility following acetate supplementation has been shown to promote cancer cell differentiation
[146].
Acetate is a nutritional source of carbon used by cancer cells for fatty acid and phospholipid biosynthesis under hypoxic conditions. A functional genomics study showed that the activity of ACSS2 contributes to cancer cell proliferation under lipid deprivation conditions
[147]. ACSS2 expression is up-regulated during metabolic stress and correlates with cancer progression and metastasis. For example, in primary and metastatic brain tumors, only 50% of carbon can be provided by glucose, whereas acetate oxidation can occur simultaneously with glucose oxidation correlating with the expression of ACSS2
[148].
Additionally, cancer cell survival and metastasis can be maintained by lipid biosynthesis promoted by a shift in glutamine metabolism from oxidation to reductive carboxylation. HIF-1 activation results in a reduction of the activity of the α-ketoglutarate dehydrogenase (α-KGDH) complex. α-KGDH catalyzes the production of succinyl-CoA and NADH from α-ketoglutarate (α-KG) and NAD
+, respectively, and is inhibited by ROS, succinyl-CoA, and an increase in NADH/NAD
+ and ATP/ADP ratios
[149]. α-KGDH is a key mitochondrial enzymatic complex in determining flux through the TCA cycle; this leads to the activation of glutamine-dependent fatty acid biosynthesis
[150].
Dietary lipids also can affect lipid metabolism and have a role in shaping the tumor micro-environment, as well as cancer progression and treatment
[151][152][151,152]. High-fat diet-induced obesity and type 2 diabetes mellitus are considered to be risk factors for many cancer types, including pancreatic and breast cancers
[153][154][153,154]. High body mass index in individuals with no previously diagnosed tumor has been shown as being associated with high cancer risk
[155]. In mice, a high-fat diet stimulates oncogenic KRAS via cyclooxygenase 2 (COX2) activation, leading to pancreatic inflammation, fibrosis, and development of invasive PDAC
[156], whereas fibroblast growth factor 21, a metabolic regulator preventing obesity, causes a reduction in tumor growth
[157].