As G protein coupled receptors, sphingosine-1-phosphate receptors (S1PRs) have recently gained attention for their role in modulating inflammatory bone loss diseases.
- bone loss
- sphingosine-1-phosphate receptors
- osteoclastogenesis
- cytokine
- chemotaxis
1. Introduction
2. Biological Effects of S1P in Inflammatory Bone Loss Diseases
2.1. S1P Biosynthesis and Degradation
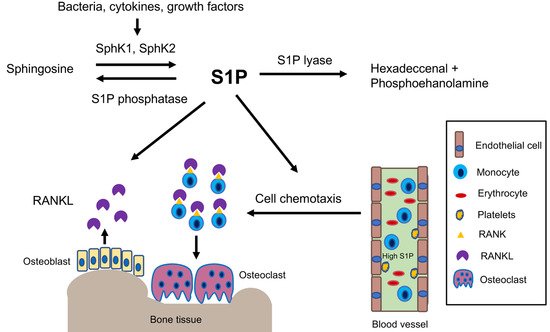
2.2. High S1P and SphK1 Levels Coincide with Inflammatory Bone Loss Diseases
2.3. Mechanisms Associated with S1P in Regulating Inflammatory Bone Loss Diseases
3. Biological Effects of S1PR2 in Inflammatory Bone Loss Diseases
3.1. Inhibition of S1PR2 by JTE013 Alleviated Inflammatory Bone Loss Diseases
S1PR2, also called endothelial differentiation G-protein coupled receptor 5 (EDG5), is located on the plasma membrane and in the cytoplasm of mammalian cells [16]. S1PR2 couples with heterotrimeric Gi, Gq, G12/13 proteins, which regulates various cellular signal- ing pathways, including adenylate cyclase, phospholipase C, (PLC), phosphoinositide-3 kinase (PI3K), nuclear kappa-B (NF-kB), extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), p38 mitogen-activated kinase (MAPK), and small G protein Rac and Rho [16][17][18][19][16–19].
The studies from our and other labs demonstrate that S1PR2 plays an essential role in modulating osteoclastogenesis and bone homeostasis. Ishii et al. [39] [39] showed that genetic deletion of S1PR2 in mice resulted in increased bone volume, numbers of trabecular bone, and trabecular thickness compared with wild type mice. They also demonstrated that pharmacological inhibition of S1PR2 by a S1PR2 inhibitor, JTE013, attenuated osteoporosis induced by RANKL [39]. In our studies, treatment with JTE013 in mice also alleviated periodontal inflammatory bone loss induced by tooth ligature placement [40]. These exper- imental data indicate that S1PR2 is a good therapeutic target for treatment of inflammatory bone loss-associated diseases.
3.2. Mechanisms Associated with S1PR2 in Regulating Inflammatory Bone Loss Diseases
3.2.1. Role of S1PR2 in Inflammatory Cytokine Release
A previous study demonstrated that the genetic deletion of S1PR2 in Apoe/S1pr2/ mice displayed reduced serum IL-1 and IL-18 levels when challenged by bacterial LPS compared to Apoe/ mice [41]. Pharmacological inhibition of S1PR2 by JTE013 in wild
type mice also reduced serum IL-1 and IL-18 levels when challenged by LPS [41]. In our studies, knockdown of S1PR2 by a S1PR2 shRNA or pharmacological inhibition of S1PR2 by JTE013 in murine BMMs decreased IL-1, IL-6, and TNF-↵ inflammatory cytokine levels that were induced by the oral bacterial pathogen A. actinomycetemcomitans [42][43][42,43]. Subse- quently, we demonstrated that treatment with either S1PR2 shRNA or JTE013 reduced PI3K, NF-kB, ERK, JNK, and p38-MAPK signaling pathways induced by A. actinomycetemcomi- tans, compared with controls [42][43][42,43]. In a ligature-induced periodontitis animal study, oral topical administration of JTE013 significantly decreased IL-1, IL-6, and TNF mRNA levels in gingival mucosa tissues when compared with a vehicle treatment group [40]. Addition- ally, in a bile duct ligation-induced cholestatic liver injury study in mice, treatment with a glucan-encapsulated S1PR2 siRNA significantly attenuated IL-1 and IL-18 in the liver, as well as serum IL-1 and IL-18 levels, compared with controls [44]. Zhao et al. [45] revealed that S1PR2 is a receptor for bile acid. Deoxycholic acid dose-dependently stimulated the up-regulation of S1PR2 [45]. Moreover, deoxycholic acid induced the generation of IL-1 in macrophages, which was blocked by inhibiting S1PR2 by JTE013 [45]. In a colitis animal study induced by deoxycholic acid and dextran sulfate sodium, treatment with JTE013 alle- viated inflammation in the colon [45]. Additionally, in an ovalbumin-induced experimental asthma study, mice treated with JTE013 exhibited significantly reduced levels of IL-4, IL-5, and IL-13 in the bronchoalveolar lavage fluid, as well as attenuated inflammation in the lungs [46]. In a bleomycin-induced lung fibrosis animal study, genetic deletion of S1PR2 in mice displayed attenuated lung fibrosis compared with wild type mice [47]. Zhao et al. [47] showed that bleomycin administration stimulated the mRNA expression of profibrotic cytokines (IL-13, and IL-4), as well as chemokine C-C motif ligand 17 (CCL17) and CCL24 in the lungs, which were markedly diminished in S1pr/ mice compared with wild type mice. Pharmacological inhibition of S1PR2 by a S1PR2 antagonist, S1PR2i, inhibited lung fibrosis induced by bleomycin [47]. These studies demonstrated that S1PR2 plays an essen- tial role in regulating the release of inflammatory cytokines and chemokines induced by various stimuli, including bacterial pathogens, LPS, bile acid, ovalbumin, and bleomycin. Interestingly, in a sepsis animal study, Hou et al. [48] demonstrated that S1PR2 also inhibits bacterial phagocytosis. Genetic deletion of S1PR2 or pharmacological inhibition of S1PR2 by JTE013 in mice reduced bacterial burden and improved survival rate in mice with sepsis by enhancing bacterial phagocytosis [48]. This was associated with S1PR2 in regulating Rac1 and filament actin (F-actin) in response to E. coli [48]. Genetic deletion of S1PR2 increased Rac1 and F-actin induced by E. coli, subsequently enhancing bacterial uptake and phagocytosis [48]. Because enhancing bacterial phagocytosis reduces the amount of bacteria in contact with immune cells and subsequently decreases the inflammatory response, the reduction of inflammatory cytokine observed in our studies [42][43][42,43] was asso- ciated with JTE013’s ability to promote bacterial phagocytosis and attenuate LPS-induced cytokine release.
3.2.2. Role of S1PR2 in Cell Chemotaxis
Previously, Ishii et al. [39] showed that murine BMMs treated with S1PR2 siRNA increased cell migration from a low concentration of S1P toward a high concentration of S1P, indicating that S1PR2 plays a chemorepulsive role induced by S1P [39]. In a RANKL- induced osteoporosis animal study, treatment with JTE013 in mice increased the number of CD11b+ monocytes in the blood compared with control [39]. In contrast, there was no significant difference in the number of CD3+ T lymphocytes in the blood between JTE013-treated mice and control [39]. This suggested that inhibition of S1PR2 by JTE013 suppressed monocytes migration from blood to bone tissues. In contrast, Yang et al. [49] showed that murine BMMs, treated with either a S1PR2 siRNA or JTE013, inhibited cell migration induced by S1P (100 nM) [49]. Using a bile duct ligation-induced cholestatic liver injury animal model, Yang et al. [49] demonstrated that mice treated with JTE013 had alleviated inflammation and fibrosis in the liver compared with controls. Treatment with JTE013 decreased the percentage of F4/80 positive macrophages in the liver compared with vehicle treatment, which also suggested that JTE013 inhibited macrophage chemotaxis from the blood circulation to the liver [49]. Because bile duct ligation stimulates the generation of various inflammatory cytokines along with S1P, both inflammatory cytokines and S1P contribute to cell chemotaxis. Therefore, it is important to evaluate the role of S1PR2 in cell chemotaxis stimulated by inflammatory cytokines. A previous study showed that treatment with a S1PR2 siRNA reduced IL-1 and IL-18 mRNA levels in liver induced by bile duct ligation [44], and treatment with JTE013 reduced IL-1 induced by deoxycholic acid [45]. Therefore, it is possible that JTE013 reduced inflammatory cytokines in the liver induced by bile duct ligation and subsequently decreased macrophage chemotaxis. In our studies, in JTE013-treated murine BMMs, we not only observed a significant reduction of levels of IL-1, IL-6, and TNF-↵ induced by the oral bacterial pathogen A. actinomycetemcomitans, we also noticed a significant reduction of S1P in murine BMMs treated with JTE013, with or without bacterial infection [43], which suggested that JTE013 down-regulates Sphk1 activity. Because treatment with JTE013 reduced both S1P and inflammatory cytokines, JTE013 inhibited monocytes chemotaxis induced by bacteria-stimulated cell culture media [43]. Treatment with S1PR2 shRNA also suppressed monocyte chemotaxis induced by bacteria-stimulated cell culture media [42][43][42,43]. In a bleomycin-induced lung fibrosis animal study, S1PR2 deficiency reduced the total number of cells and the number of macrophages in the bronchoalveolar lavage fluid [47]. These studies demonstrated that S1PR2 controls inflammatory cytokine and/or S1P release, subsequently affecting inflammatory cell chemotaxis. S1PR2 deficiency or pharmacological inhibition of S1PR2 by JTE013 inhibited cell chemotaxis induced by various stimuli.
3.2.3. Role of S1PR2 in RANKL-Induced Osteoclastogenesis
A previous study [39] in mice showed that pharmacological inhibition of S1PR2 by JTE013 suppressed RANKL-induced osteoporosis. Our studies revealed that S1PR2 controls cell adhesion units (podosomes) induced by RANKL in BMMs, which influence both osteoclastogenesis and bone resorption. Podosomes are basic cell adhesion units that are required for cell adhesion and fusion to form multinucleated osteoclasts [50][51][52][50–52]. RANKL stimulated the up-regulation of podosome components (including PI3K, Src, Pyk2, F-actin, integrin 3, and paxillin levels), which were suppressed by treatment with either S1PR2 shRNA or JTE013 compared with controls [43]. This effect was not associated with the S1P signal, since S1P has no effect on the differentiation of osteoclasts in the single culture of BMMs [35]. Treatment with S1PR2 shRNA or JTE013 in BMMs significantly decreased various osteoclastogenic genes, including nuclear factor of activated T-cells cytoplasmic calcineurin-dependent 1 (Nfatc1), cathepsin K (Ctsk), acid phosphatase 5 (Acp5), osteoclast- associated receptor (Oscar), dendritic cell-specific transmembrane protein (Dc-stamp), and osteoclast stimulatory transmembrane protein (Oc-stamp) induced by RANKL compared with controls [42][43][42,43]. Using a ligature placement induced periodontitis animal model, we demonstrated that treatment with JTE013 reduced the number of osteoclasts in the periodontal tissues and attenuated alveolar bone loss [40]. Our study demonstrated that S1PR2 plays a key role in regulating RANKL-induced osteoclastogenesis. Treatment with JTE013 inhibits inflammatory bone loss by multiple mechanisms, including suppressing the production of inflammatory cytokines and S1P, reducing monocyte chemotaxis, and inhibiting RANKL-induced adhesion and fusion of osteoclast precursors (Figure 2).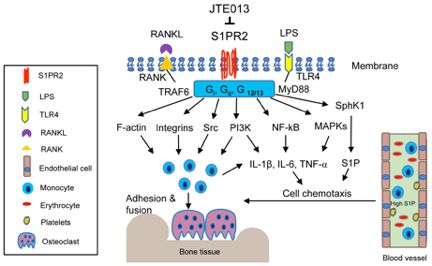 Figure 2. Simplified illustration of biological effects of S1PR2 in inflammatory bone loss diseases. S1PR2 couples with Gi, Gq, and G12/13 proteins, which manipulate multiple signaling pathways affecting inflammatory diseases. First, S1PR2 controls PI3K, NF-kB, and MAPKs signaling pathways induced by LPS, influencing IL-1, IL-6, TNF-↵ inflammatory cytokine release. Second, S1PR2 modulates Sphk1 and S1P generation induced by inflammation. Third, S1PR2 manipulates chemotaxis of osteoclast precursors from blood circulation to bone and soft tissues by modulating inflammatory cytokine and S1P generation. Fourth, S1PR2 controls podosome components (F-actin, integrins, Src, PI3K) induced by RANKL and modulates the adhesion and fusion of osteoclast precursors. S1PR2 possibly interacts with Gi, Gq, and G12/13 proteins, RANKL adaptor protein TRAF6, and TLR4 adaptor protein MyD88 in lipid rafts after stimulation by RANKL or LPS.
Figure 2. Simplified illustration of biological effects of S1PR2 in inflammatory bone loss diseases. S1PR2 couples with Gi, Gq, and G12/13 proteins, which manipulate multiple signaling pathways affecting inflammatory diseases. First, S1PR2 controls PI3K, NF-kB, and MAPKs signaling pathways induced by LPS, influencing IL-1, IL-6, TNF-↵ inflammatory cytokine release. Second, S1PR2 modulates Sphk1 and S1P generation induced by inflammation. Third, S1PR2 manipulates chemotaxis of osteoclast precursors from blood circulation to bone and soft tissues by modulating inflammatory cytokine and S1P generation. Fourth, S1PR2 controls podosome components (F-actin, integrins, Src, PI3K) induced by RANKL and modulates the adhesion and fusion of osteoclast precursors. S1PR2 possibly interacts with Gi, Gq, and G12/13 proteins, RANKL adaptor protein TRAF6, and TLR4 adaptor protein MyD88 in lipid rafts after stimulation by RANKL or LPS.
3.2.4. Possible Interaction of S1PR2 with Other Signaling Molecules in Lipid Rafts
It is well known that mammalian membrane contains specialized membrane domains, called lipid rafts, which are enriched in cholesterol, glycosphingolipid, and proteins. Lipids rafts serve as signaling platforms that recruit transmembrane and intracellular signaling molecules, facilitating the interaction of signaling molecules and signaling transduction following various stimuli [53][54][53,54]. Heterotrimeric G proteins, Src, PI3K, integrins, and MAPKs are some of the signaling molecules within the lipid rafts [53][54][53,54]. Additionally, it has been shown that toll-like receptor 4 (TLR4) and its adaptor protein MyD88 are recruited to the lipid rafts in response to oxidase stress or fatty acid treatment [55][56][55,56]. RANK and its adaptor protein TRAF6 are also recruited to lipid rafts after RANKL stimulation [57]. There- fore, it is possible that S1PR2 might interact with heterotrimeric Gi, Gq, G12/13 proteins, TLR4, MyD88, RANK, TRAF6, Src, PI3K, integrins, and MAPKs within the lipid rafts after bacterial LPS or RANKL stimulation. Interestingly, it has been reported that treatment with vitamin D and its analog, eldecalcitol, reduced S1PR2 mRNA levels in murine circulating osteoclast precursors and alleviated ovariectomy-induced osteoporosis [58]. Additionally, incubation of murine bone marrow cells with IL-6 increased S1PR2 mRNA levels [59]. Treatment with an IL-6 receptor antibody alleviated collagen-induced bone loss by reduc- ing S1PR2 in bone marrow cells [59]. These data support our hypothesis that S1PR2, as a G-protein coupled receptor, not only modulates S1P signaling, but also controls other stimuli, including bacterial LPS, RANKL, bile acid, vitamin D, and IL-6 [42][43][48][58][59][42,43,48,58,59]. It is likely that S1PR2 interacts with various signaling molecules in the lipid rafts, subsequently modulating bone homeostasis.
4. Biological Effects of a S1PRs Modulator, FTR720, in Inflammatory Bone Loss Diseases
4.1. FTY720 Attenuated Inflammatory Bone Loss in Animal Studies
FTY720, also called fingolimod, is a S1PRs modulator. FTY720 is synthesized by structural modification of myriocin, a fungal metabolite from Isaclaria sinclarii, a traditional herb used in Eastern medicine [60]. FTY720 is phosphorylated to p-FTY720 by SphK2. Ini- tially, FTY720 was considered a multiple S1PRs (including S1PR1, S1PR3-5) agonist [61][62][61,62]. However, later studies discovered that FTY720 also functions as a noncompetitive inhibitor of multiple S1PRs by promoting internalization and partial degradation of S1PRs [63][64][63,64]. In previous in vitro studies, FTY720 inhibited the binding of S1P to S1PR1, S1PR5, and to a lesser extent, S1PR2 in lymphocytes [63]. Additionally, FTY720 reduced S1PR1 and S1PR4 levels in dendritic cells [64]. FTY720 has been used in clinical trials as an immune suppressant to treat patients with autoimmune diseases, including relapsing multiple scle- rosis, as well as in patients with renal transplant to prevent rejection of the transplant [65] [65]. Additionally, FTY720 exhibits an anti-inflammatory bone loss effect in animals. Treat- ment with FTY720 in mice alleviated ovariectomy-induced osteoporosis [66][67][66,67], attenuated apical periodontitis [31], and suppressed bone destruction and hindpaw edema in animals with arthritis induced either by collagen, adjuvant, or an arthrogenic anti-collagen II antibody [67][68][69][70][71].
mals with arthritis induced either by collagen, adjuvant, or an arthrogenic anti-collagen II antibody [67–71].
4.2. Mechanisms Associated with FTY720 in Modulating Inflammatory Bone Loss Diseases
4.2.1. Role of FTY720 in Inflammatory Cytokine Response
We previously reported that treatment with FTY720 (2 to 8 μM) in murine BMMs dose-dependently reduced IL-1, IL-6, and TNF-α induced by the oral bacterial pathogen A. actinomycetemcomitans by inhibiting PI3K, ERK, and Akt signaling pathways [72]. FTY720 also suppressed IL-1, IL-6, and TNF-↵ induced by LPS in microglia [73], and reduced IL-6, IL-12, TNF-α, and MCP-1 induced by LPS in bone marrow-derived dendritic cells (BMDCs) [74]. Zeng et al. [74] revealed that the anti-inflammatory effect of FTY720 was associated with FTY720 in altering cell shape, surface markers, and antigen presentation induced by LPS in BMDCs [74]. Upon LPS stimulation, the shape of BMDCs became elon- gated. This elongation of BMDCs was suppressed by treating BMDCs with FTY720 [74]. After stimulation by microbial products or cytokines, immature dendritic cells acquire certain surface markers (including MHC II and co-stimulatory molecules), leading to phenotypic and functional maturation processes. Zeng et al. [74] showed that LPS in- duced up-regulation of surface markers (including MHC II molecule, I-Ad, co-stimulatory molecule CD86, and adhesion molecule CD40) that were suppressed by treatment with FTY720 [74]. In a collagen-induced arthritis animal model, FTY720-treated mice displayed reduced IL-1, IL-6, and TNF-α in plasma and decreased IL-6, and TNF-α in synovial tissues [69]. Additionally, FTY720 altered cytokine profiles in dendritic cells [64]. Mature dendritic cells generate distinct Th lineage-polarizing cytokines that induce clonal expan- sion of naïve T cells and initiate a primary adaptive immune response. Muller et al. [64] showed that co-culturing of naïve T cells with FTY720 or p-FTY720-treated dendritic cells enhanced the production of IL-4, but reduced the production of IFN-, suggesting that FTY720 promoted the shift from the Th1 to Th2 cytokine profile. Moreover, high concen- tration (3 to 10 μM) of FTY720 can also increase cell membrane permeability and promote necrosis of inflammatory cells [75], which in turn decrease inflammatory cytokine pro- duction. Furthermore, in a collagen-induced arthritis animal study, FTY720 also reduced an anti-type II collagen antibody [70]. These in vitro and in vivo studies demonstrated FTY7200s ability to inhibit inflammatory cytokine production, alter cytokine Th profiles, and suppress antibody production.
4.2.2. Role of FTY720 in Regulating Cell Migration and Chemotaxis
In animals treated with FTY720, the number of lymphocytes was markedly decreased in the peripheral blood, thoracic lymph duct, and partially in the spleen. In contrast, the number of lymphocytes in peripheral lymph nodes, mesenteric lymph nodes, and Peyer’s patch was significantly increased [76]. This effect was caused by degradation of S1PR1 by FTY720, which reduces the adhesion of T cells on the lymph node sinus and suppresses the egress of lymphocytes from the secondary lymphoid organs to lymph and peripheral blood, leading to lymphopenia [77][78][79][77–79]. In adjuvant-induced arthritis studies, FTY720 treatment (0.03 to 0.3 mg/kg) significantly reduced the number of lymphocytes in the peripheral blood, without affecting the number of leukocytes and monocytes in the blood [70][71][70,71]. In a collagen-induced arthritis study, FTY720-treated animals had reduced CD4+ T cell infiltration in the synovium [68]. Additionally, FTY720 controls dendritic cell migration induced by chemokines [64] and modulates dendritic cell migration from peripheral tissues to draining lymph nodes [69]. Muller et al. [64] revealed that dendritic cells treated with either FTY720 or p-FTY720 diminished dendritic cells chemotaxis in response to chemokines (RANTES or SDF-1α). This effect was associated with a reduction of F-actin after treatment with FTY720 or p-FTY72 in BMDCs [64]. Dendritic cells serve as sentinels in the immune system by capturing and processing antigens at peripheral tissues. After activation of immature dendritic cells by microbial products or inflammatory cytokines, dendritic cells undergo cell maturation and migration to draining lymph nodes where dendritic cell interact with naïve T lymphocytes and convert them into antigen-specific reactive T cells. In a collagen-induced arthritis study in mice [69], treatment with FTY720 (5 mg/kg) inhibited dendritic cell migration from peripheral tissues to draining lymph nodes. This was associated with inhibiting production of chemokine CCL19 and chemokine receptor CCR7 in dendritic cells treated with FTY720 [69]. In other collagen-induced arthritis studies in mice [68][70], treatment with FTY720 (0.3 or 0.6 mg/kg) reduced the number of both lymphocytes and monocytes in the blood. Because FTY720 suppresses inflammatory cytokine release in tissues, FTY720 could reduce the chemotaxis of various inflammatory cells (including T cells, dendritic cells, and monocytes) from blood to peripheral tissues.
interact with naïve T lymphocytes and convert them into antigen-specific reactive T cells. In a collagen-induced arthritis study in mice [69], treatment with FTY720 (5 mg/kg) inhibited dendritic cell migration from peripheral tissues to draining lymph nodes. This was associated with inhibiting production of chemokine CCL19 and chemokine receptor CCR7 in dendritic cells treated with FTY720 [69]. In other collagen-induced arthritis studies in mice [68,70], treatment with FTY720 (0.3 or 0.6 mg/kg) reduced the number of both lymphocytes and monocytes in the blood. Because FTY720 suppresses inflammatory cytokine release in tissues, FTY720 could reduce the chemotaxis of various inflammatory cells (including T cells, dendritic cells, and monocytes) from blood to peripheral tissues.
4.2.3. Role of FTY720 in RANKL-Induced Osteoclastogenesis
We have demonstrated that FTY720 suppressed osteoclastogenesis induced by RANKL in murine bone marrow cells [72]. Treatment with FTY720 decreased osteoclastogenic genes, including Nfactc1, Ctsk, Acp5, and Oscar induced by RANKL, compared with vehicle treatment [72]. Our study suggests that FTY720 modulates cell signaling pathways induced by RANKL. Additionally, reducing the number of infiltrated T cells in the bone and sur- rounding soft tissues by FTY720 could further decrease RANKL production, subsequently suppressing osteoclastogenesis. In an apical periodontitis animal study, the number of osteoclasts and RANKL expression decreased significantly in the periodontal tissues of the FTY720-treated group compared with the control group [31]. In summary, studies from our and other labs support that FTY720 attenuates inflammatory bone loss by reducing inflammatory cytokine release, inhibiting inflammatory cell migration from blood to bone and surrounding soft tissues, and suppressing osteoclastogenesis induced by RANKL (Figure 3).
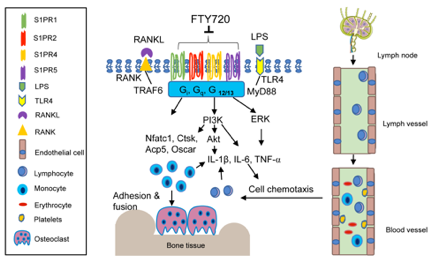
Figure 3. Simplified illustration of biological effect of FTY720 in inflammatory bone loss diseases. FTY720 can internalize and partially degrade multiple S1PRs, including S1PR1, S1PR2, S1PR4, and S1PR5. FTY720 inhibits IL-1, IL-6, and TNF-↵ inflammatory cytokine release by blocking PI3K, Akt, and ERK signaling pathways. FTY720 suppresses T lymphocyte egress from draining lymph nodes to the lymph and peripheral blood. Reduction of inflammatory cytokines and the number of T lymphocytes in the blood subsequently decreases monocytes and T lymphocytes in the bone and soft tissues. Additionally, FTY720 inhibits Nfact1, Ctsk, Acp5, and Oscar genes, suppressing RANKL-induced osteoclastogenesis.
References
- Redlich, K.; Smolen, J. S., Inflammatory bone loss: pathogenesis and therapeutic intervention. Nat Rev Drug Discov 2012, 11, (3), 234-50.
- Vaananen, H. K.; Laitala-Leinonen, T., Osteoclast lineage and function. Arch Biochem Biophys 2008, 473, (2), 132-8.
- Ono, T.; Nakashima, T., Recent advances in osteoclast biology. Histochemistry and cell biology 2018, 149, (4), 325-341.
- Weitzmann, M. N., The Role of Inflammatory Cytokines, the RANKL/OPG Axis, and the Immunoskeletal Interface in Physiological Bone Turnover and Osteoporosis. Scientifica (Cairo) 2013, 2013, 125705.
- Walsh, M. C.; Choi, Y., Biology of the RANKL-RANK-OPG System in Immunity, Bone, and Beyond. Frontiers in immunology 2014, 5, 511.
- Vaananen, K., Mechanism of osteoclast mediated bone resorption--rationale for the design of new therapeutics. Adv Drug Deliv Rev 2005, 57, (7), 959-71.
- Hienz, S. A.; Paliwal, S.; Ivanovski, S., Mechanisms of Bone Resorption in Periodontitis. J Immunol Res 2015,2015:615486., (doi), 10.1155/2015/615486. Epub 2015 May 3.
- Pacifici, R., Estrogen deficiency, T cells and bone loss. Cell Immunol. 2008, 252, (1-2), 68-80. doi: 10.1016/j.cellimm.2007.06.008. Epub 2007 Sep 20.
- Yokota, K., [Inflammation and osteoclasts]. Nihon Rinsho Meneki Gakkai Kaishi 2017, 40, (5), 367-376. doi: 10.2177/jsci.40.367.
- Kwan Tat, S.; Padrines, M.; Théoleyre, S.; Heymann, D.; Fortun, Y., IL-6, RANKL, TNF-alpha/IL-1: interrelations in bone resorption pathophysiology. Cytokine Growth Factor Rev. 2004, 15, (1), 49-60. doi: 10.1016/j.cytogfr.2003.10.005.
- Marahleh, A.; Kitaura, H.; Ohori, F.; Kishikawa, A.; Ogawa, S.; Shen, W. R.; Qi, J.; Noguchi, T.; Nara, Y.; Mizoguchi, I., TNF-α Directly Enhances Osteocyte RANKL Expression and Promotes Osteoclast Formation. Front Immunol. 2019, 10:2925., (doi), 10.3389/fimmu.2019.02925. eCollection 2019.
- Baum, R.; Gravallese, E. M., Impact of inflammation on the osteoblast in rheumatic diseases. Curr Osteoporos Rep 2014, 12, (1), 9-16.
- Kotake, S.; Nanke, Y., Effect of TNFalpha on osteoblastogenesis from mesenchymal stem cells. Biochimica et biophysica acta 2014, 1840, (3), 1209-13.
- Matsugaki, A.; Kimura, Y.; Watanabe, R.; Nakamura, F.; Takehana, R.; Nakano, T., Impaired Alignment of Bone Matrix Microstructure Associated with Disorganized Osteoblast Arrangement in Malignant Melanoma Metastasis. Biomolecules 2021, 11, (2).
- Matsugaki, A.; Matsumoto, S.; Nakano, T., A Novel Role of Interleukin-6 as a Regulatory Factor of Inflammation-Associated Deterioration in Osteoblast Arrangement. International journal of molecular sciences 2020, 21, (18).
- Aarthi, J. J.; Darendeliler, M. A.; Pushparaj, P. N., Dissecting the role of the S1P/S1PR axis in health and disease. J Dent Res 2011, 90, (7), 841-54.
- Siehler, S.; Manning, D. R., Pathways of transduction engaged by sphingosine 1-phosphate through G protein-coupled receptors. Biochimica et biophysica acta 2002, 1582, (1-3), 94-9.
- Takuwa, Y., Subtype-specific differential regulation of Rho family G proteins and cell migration by the Edg family sphingosine-1-phosphate receptors. Biochim Biophys Acta. 2002, 1582, (1-3), 112-20.
- Kluk, M. J.; Hla, T., Signaling of sphingosine-1-phosphate via the S1P/EDG-family of G-protein-coupled receptors. Biochim Biophys Acta. 2002, 1582, (1-3), 72-80.
- El Alwani, M.; Wu, B. X.; Obeid, L. M.; Hannun, Y. A., Bioactive sphingolipids in the modulation of the inflammatory response. Pharmacol Ther 2006, 112, (1), 171-83.
- Rivera, J.; Proia, R. L.; Olivera, A., The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat Rev Immunol 2008, 8, (10), 753-63.
- Snider, A. J.; Orr Gandy, K. A.; Obeid, L. M., Sphingosine kinase: Role in regulation of bioactive sphingolipid mediators in inflammation. Biochimie 2010, 92, (6), 707-15.
- Pitson, S. M., Regulation of sphingosine kinase and sphingolipid signaling. Trends Biochem Sci 2011, 36, (2), 97-107.
- Wattenberg, B. W., Role of sphingosine kinase localization in sphingolipid signaling. World J Biol Chem 2010,1, (12), 362-8.
- Xia, P.; Wadham, C., Sphingosine 1-phosphate, a key mediator of the cytokine network: juxtacrine signaling. Cytokine Growth Factor Rev 2011, 22, (1), 45-53.
- Yu, H.; Sun, C.; Argraves, K. M., Periodontal inflammation and alveolar bone loss induced by Aggregatibacter actinomycetemcomitans is attenuated in sphingosine kinase 1-deficient mice. J Periodontal Res 2016, 51, (1), 38-49.
- von Wenckstern, H.; Zimmermann, K.; Kleuser, B., The role of the lysophospholipid sphingosine 1-phosphate in immune cell biology. Arch Immunol Ther Exp (Warsz) 2006, 54, (4), 239-51.
- Spiegel, S.; Milstien, S., The outs and the ins of sphingosine-1-phosphate in immunity. Nat Rev Immunol 2011,11, (6), 403-15.
- Lee, S. H.; Lee, S. Y.; Lee, Y. S.; Kim, B. J.; Lim, K. H.; Cho, E. H.; Kim, S. W.; Koh, J. M.; Kim, G. S., Higher circulating sphingosine 1-phosphate levels are associated with lower bone mineral density and higher bone resorption marker in humans. J Clin Endocrinol Metab 2012, 97, (8), E1421-8.
- Lai, W. Q.; Chia, F. L.; Leung, B. P., Sphingosine kinase and sphingosine-1-phosphate receptors: novel therapeutic targets of rheumatoid arthritis? Future Med Chem 2012, 4, (6), 727-33.
- Xiao, L.; Zhou, Y.; Zhu, L.; Yang, S.; Huang, R.; Shi, W.; Peng, B.; Xiao, Y., SPHK1-S1PR1-RANKL Axis Regulates the Interactions Between Macrophages and BMSCs in Inflammatory Bone Loss. J Bone Miner Res. 2018, 33, (6), 1090-1104. doi: 10.1002/jbmr.3396. Epub 2018 Feb 21.
- Baker, D. A.; Barth, J.; Chang, R.; Obeid, L. M.; Gilkeson, G. S., Genetic sphingosine kinase 1 deficiency significantly decreases synovial inflammation and joint erosions in murine TNF-alpha-induced arthritis. Journal of immunology (Baltimore, Md. : 1950) 2010, 185, (4), 2570-9.
- Jougasaki, M.; Ichiki, T.; Takenoshita, Y.; Setoguchi, M., Statins suppress interleukin-6-induced monocyte chemo-attractant protein-1 by inhibiting Janus kinase/signal transducers and activators of transcription pathways in human vascular endothelial cells. Br J Pharmacol 2010, 159, (6), 1294-303.
- Rider, P.; Carmi, Y.; Guttman, O.; Braiman, A.; Cohen, I.; Voronov, E.; White, M. R.; Dinarello, C. A.; Apte, R. N., IL-1alpha and IL-1beta recruit different myeloid cells and promote different stages of sterile inflammation. Journal of immunology (Baltimore, Md. : 1950) 2011, 187, (9), 4835-43.
- Ryu, J.; Kim, H. J.; Chang, E. J.; Huang, H.; Banno, Y.; Kim, H. H., Sphingosine 1-phosphate as a regulator of osteoclast differentiation and osteoclast-osteoblast coupling. EMBO J 2006, 25, (24), 5840-51.
- Quint, P.; Ruan, M.; Pederson, L.; Kassem, M.; Westendorf, J. J.; Khosla, S.; Oursler, M. J., Sphingosine 1-phosphate (S1P) receptors 1 and 2 coordinately induce mesenchymal cell migration through S1P activation of complementary kinase pathways. J Biol Chem 2013, 288, (8), 5398-406.
- Matsuzaki, E.; Hiratsuka, S.; Hamachi, T.; Takahashi-Yanaga, F.; Hashimoto, Y.; Higashi, K.; Kobayashi, M.; Hirofuji, T.; Hirata, M.; Maeda, K., Sphingosine-1-phosphate promotes the nuclear translocation of beta-catenin and thereby induces osteoprotegerin gene expression in osteoblast-like cell lines. Bone 2013, 55, (2), 315-24.
- Weske, S.; Vaidya, M.; Reese, A.; von Wnuck Lipinski, K.; Keul, P.; Bayer, J. K.; Fischer, J. W.; Flogel, U.; Nelsen, J.; Epple, M.; Scatena, M.; Schwedhelm, E.; Dorr, M.; Volzke, H.; Moritz, E.; Hannemann, A.; Rauch, B. H.; Graler, M. H.; Heusch, G.; Levkau, B., Targeting sphingosine-1-phosphate lyase as an anabolic therapy for bone loss. Nat Med 2018, 24, (5), 667-678.
- Ishii, M.; Kikuta, J.; Shimazu, Y.; Meier-Schellersheim, M.; Germain, R. N., Chemorepulsion by blood S1P regulates osteoclast precursor mobilization and bone remodeling in vivo. J Exp Med 2010, 207, (13), 2793-8.
- Snipes, M.; Sun, C.; Yu, H., Inhibition of sphingosine-1-phosphate receptor 2 attenuated ligature-induced periodontitis in mice. Oral diseases 2020.
- Skoura, A.; Michaud, J.; Im, D. S.; Thangada, S.; Xiong, Y.; Smith, J. D.; Hla, T., Sphingosine-1-phosphate receptor-2 function in myeloid cells regulates vascular inflammation and atherosclerosis. Arterioscler Thromb Vasc Biol. 2011, 31, (1), 81-5.
- Yu, H., Sphingosine-1-Phosphate Receptor 2 Regulates Proinflammatory Cytokine Production and Osteoclastogenesis. PloS one 2016, 11, (5), e0156303.
- Hsu, L. C.; Reddy, S. V.; Yilmaz, O.; Yu, H., Sphingosine-1-Phosphate Receptor 2 Controls Podosome Components Induced by RANKL Affecting Osteoclastogenesis and Bone Resorption. Cells 2019, 8, (1).
- Hou, L.; Yang, L.; Chang, N.; Zhao, X.; Zhou, X.; Dong, C.; Liu, F.; Yang, L.; Li, L., Macrophage Sphingosine 1-Phosphate Receptor 2 Blockade Attenuates Liver Inflammation and Fibrogenesis Triggered by NLRP3 Inflammasome. Frontiers in immunology 2020, 11, 1149.
- Zhao, S.; Gong, Z.; Du, X.; Tian, C.; Wang, L.; Zhou, J.; Xu, C.; Chen, Y.; Cai, W.; Wu, J., Deoxycholic Acid-Mediated Sphingosine-1-Phosphate Receptor 2 Signaling Exacerbates DSS-Induced Colitis through Promoting Cathepsin B Release. J Immunol Res 2018, 2018, 2481418.
- Terashita, T.; Kobayashi, K.; Nagano, T.; Kawa, Y.; Tamura, D.; Nakata, K.; Yamamoto, M.; Tachihara, M.; Kamiryo, H.; Nishimura, Y., Administration of JTE013 abrogates experimental asthma by regulating proinflammatory cytokine production from bronchial epithelial cells. Respir Res 2016, 17, (1), 146.
- Zhao, J.; Okamoto, Y.; Asano, Y.; Ishimaru, K.; Aki, S.; Yoshioka, K.; Takuwa, N.; Wada, T.; Inagaki, Y.; Takahashi, C.; Nishiuchi, T.; Takuwa, Y., Sphingosine-1-phosphate receptor-2 facilitates pulmonary fibrosis through potentiating IL-13 pathway in macrophages. PloS one 2018, 13, (5), e0197604.
- Hou, J.; Chen, Q.; Zhang, K.; Cheng, B.; Xie, G.; Wu, X.; Luo, C.; Chen, L.; Liu, H.; Zhao, B.; Dai, K.; Fang, X., Sphingosine 1-phosphate Receptor 2 Signaling Suppresses Macrophage Phagocytosis and Impairs Host Defense against Sepsis. Anesthesiology 2015, 123, (2), 409-22.
- Yang, L.; Han, Z.; Tian, L.; Mai, P.; Zhang, Y.; Wang, L.; Li, L., Sphingosine 1-Phosphate Receptor 2 and 3 Mediate Bone Marrow-Derived Monocyte/Macrophage Motility in Cholestatic Liver Injury in Mice. Sci Rep2015, 5, 13423.
- Georgess, D.; Machuca-Gayet, I.; Blangy, A.; Jurdic, P., Podosome organization drives osteoclast-mediated bone resorption. Cell Adh Migr 2014, 8, (3), 191-204.
- Duong, L. T.; Lakkakorpi, P.; Nakamura, I.; Rodan, G. A., Integrins and signaling in osteoclast function. Matrix Biol 2000, 19, (2), 97-105.
- Feng, X.; Teitelbaum, S. L., Osteoclasts: New Insights. Bone Res 2013, 1, (1), 11-26.
- Pike, L. J., Lipid rafts: bringing order to chaos. J Lipid Res 2003, 44, (4), 655-67.
- Simons, K.; Toomre, D., Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 2000, 1, (1), 31-9.
- Powers, K. A.; Szaszi, K.; Khadaroo, R. G.; Tawadros, P. S.; Marshall, J. C.; Kapus, A.; Rotstein, O. D., Oxidative stress generated by hemorrhagic shock recruits Toll-like receptor 4 to the plasma membrane in macrophages. J Exp Med 2006, 203, (8), 1951-61
- Wong, S. W.; Kwon, M. J.; Choi, A. M.; Kim, H. P.; Nakahira, K.; Hwang, D. H., Fatty acids modulate Toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J Biol Chem 2009, 284, (40), 27384-92
- Ha, H.; Kwak, H. B.; Le, S. W.; Kim, H. H.; Lee, Z. H., Lipid rafts are important for the association of RANK and TRAF6. Exp Mol Med 2003, 35, (4), 279-84.
- Kikuta, J.; Kawamura, S.; Okiji, F.; Shirazaki, M.; Sakai, S.; Saito, H.; Ishii, M., Sphingosine-1-phosphate-mediated osteoclast precursor monocyte migration is a critical point of control in antibone-resorptive action of active vitamin D. Proc Natl Acad Sci U S A 2013, 110, (17), 7009-13.
- Tanaka, K.; Hashizume, M.; Mihara, M.; Yoshida, H.; Suzuki, M.; Matsumoto, Y., Anti-interleukin-6 receptor antibody prevents systemic bone mass loss via reducing the number of osteoclast precursors in bone marrow in a collagen-induced arthritis model. Clin Exp Immunol. 2014, 175, (2), 172-80.
- Fujita, T.; Inoue, K.; Yamamoto, S.; Ikumoto, T.; Sasaki, S.; Toyama, R.; Chiba, K.; Hoshino, Y.; Okumoto, T., Fungal metabolites. Part 11. A potent immunosuppressive activity found in Isaria sinclairii metabolite. J Antibiot (Tokyo) 1994, 47, (2), 208-15.
- Brinkmann, V.; Davis, M. D.; Heise, C. E.; Albert, R.; Cottens, S.; Hof, R.; Bruns, C.; Prieschl, E.; Baumruker, T.; Hiestand, P.; Foster, C. A.; Zollinger, M.; Lynch, K. R., The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem 2002, 277, (24), 21453-7
- Maceyka, M.; Harikumar, K. B.; Milstien, S.; Spiegel, S., Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol 2012, 22, (1), 50-60.
- Graler, M. H.; Goetzl, E. J., The immunosuppressant FTY720 down-regulates sphingosine 1-phosphate G-protein-coupled receptors. FASEB J. 2004, 18, (3), 551-3.
- Muller, H.; Hofer, S.; Kaneider, N.; Neuwirt, H.; Mosheimer, B.; Mayer, G.; Konwalinka, G.; Heufler, C.; Tiefenthaler, M., The immunomodulator FTY720 interferes with effector functions of human monocyte-derived dendritic cells. Eur J Immunol 2005, 35, (2), 533-45.
- Montalban, X.; Comi, G.; O'Connor, P.; Gold, S.; de Vera, A.; Eckert, B.; Kappos, L., Oral fingolimod (FTY720) in relapsing multiple sclerosis: impact on health-related quality of life in a phase II study. Mult Scler 2011, 17, (11), 1341-50.
- Ishii, M.; Egen, J. G.; Klauschen, F.; Meier-Schellersheim, M.; Saeki, Y.; Vacher, J.; Proia, R. L.; Germain, R. N., Sphingosine-1-phosphate mobilizes osteoclast precursors and regulates bone homeostasis. Nature 2009, 458, (7237), 524-8.
- Kikuta, J.; Iwai, K.; Saeki, Y.; Ishii, M., S1P-targeted therapy for elderly rheumatoid arthritis patients with osteoporosis. Rheumatol Int 2011, 31, (7), 967-9.
- Wang, F.; Tan, W.; Guo, D.; He, S., Reduction of CD4 positive T cells and improvement of pathological changes of collagen-induced arthritis by FTY720. European journal of pharmacology 2007, 573, (1-3), 230-40.
- Han, Y.; Li, X.; Zhou, Q.; Jie, H.; Lao, X.; Han, J.; He, J.; Liu, X.; Gu, D.; He, Y.; Sun, E., FTY720 Abrogates Collagen-Induced Arthritis by Hindering Dendritic Cell Migration to Local Lymph Nodes. Journal of immunology (Baltimore, Md. : 1950) 2015, 195, (9), 4126-35.
- Matsuura, M.; Imayoshi, T.; Okumoto, T., Effect of FTY720, a novel immunosuppressant, on adjuvant- and collagen-induced arthritis in rats. Int J Immunopharmacol 2000, 22, (4), 323-31.
- Matsuura, M.; Imayoshi, T.; Chiba, K.; Okumoto, T., Effect of FTY720, a novel immunosuppressant, on adjuvant-induced arthritis in rats. Inflamm Res 2000, 49, (8), 404-10.
- Yu, H.; Herbert, B. A.; Valerio, M.; Yarborough, L.; Hsu, L. C.; Argraves, K. M., FTY720 inhibited proinflammatory cytokine release and osteoclastogenesis induced by Aggregatibacter actinomycetemcomitans. Lipids Health Dis 2015, 14, (1), 66.
- Noda, H.; Takeuchi, H.; Mizuno, T.; Suzumura, A., Fingolimod phosphate promotes the neuroprotective effects of microglia. J Neuroimmunol. 2013, 256, (1-2), 13-8. .
- Zeng, X.; Wang, T.; Zhu, C.; Xing, X.; Ye, Y.; Lai, X.; Song, B.; Zeng, Y., Topographical and biological evidence revealed FTY720-mediated anergy-polarization of mouse bone marrow-derived dendritic cells in vitro. PloS one 2012, 7, (5), e34830.
- Oyama, Y.; Chikahisa, L.; Kanemaru, K.; Nakata, M.; Noguchi, S.; Nagano, T.; Okazaki, E.; Hirata, A., Cytotoxic actions of FTY720, a novel immunosuppressant, on thymocytes and brain neurons dissociated from the rat. Japanese journal of pharmacology 1998, 76, (4), 377-85.
- Chiba, K.; Yanagawa, Y.; Masubuchi, Y.; Kataoka, H.; Kawaguchi, T.; Ohtsuki, M.; Hoshino, Y., FTY720, a novel immunosuppressant, induces sequestration of circulating mature lymphocytes by acceleration of lymphocyte homing in rats. I. FTY720 selectively decreases the number of circulating mature lymphocytes by acceleration of lymphocyte homing. Journal of immunology (Baltimore, Md. : 1950) 1998, 160, (10), 5037-44.
- Matloubian, M.; Lo, C. G.; Cinamon, G.; Lesneski, M. J.; Xu, Y.; Brinkmann, V.; Allende, M. L.; Proia, R. L.; Cyster, J. G., Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, (6972), 355-60.
- Chiba, K.; Adachi, K., Discovery of fingolimod, the sphingosine 1-phosphate receptor modulator and its application for the therapy of multiple sclerosis. Future Med Chem 2012, 4, (6), 771-81.
- Zhi, L.; Kim, P.; Thompson, B. D.; Pitsillides, C.; Bankovich, A. J.; Yun, S. H.; Lin, C. P.; Cyster, J. G.; Wu, M. X., FTY720 blocks egress of T cells in part by abrogation of their adhesion on the lymph node sinus. J 187, (5), 2244-51 doi: 10 4049/jimmunol 1100670 Epub 2011 Jul 25.