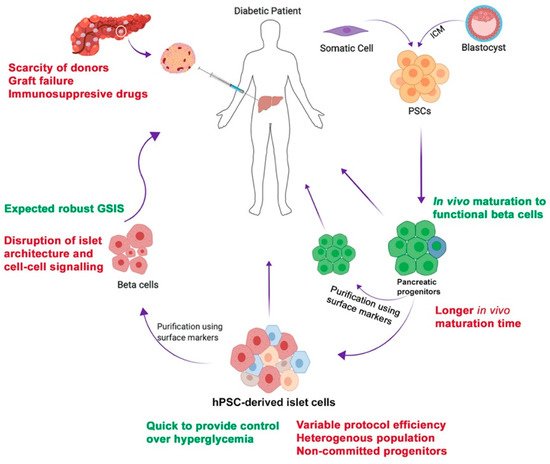
Diabetes mellitus (DM) is one of the most prevalent metabolic disorders. In order to replace the function of the destroyed pancreatic beta cells in diabetes, islet transplantation is the most widely practiced treatment. However, it has several limitations. As an alternative approach, human pluripotent stem cells (hPSCs) can provide an unlimited source of pancreatic cells that have the ability to secrete insulin in response to a high blood glucose level. However, the determination of the appropriate pancreatic lineage candidate for the purpose of cell therapy for the treatment of diabetes is still debated. While hPSC-derived beta cells are perceived as the ultimate candidate, their efficiency needs further improvement in order to obtain a sufficient number of glucose responsive beta cells for transplantation therapy. On the other hand, hPSC-derived pancreatic progenitors can be efficiently generated in vitro and can further mature into glucose responsive beta cells in vivo after transplantation.
- hPSCs
- hyperglycemia
- insulin-secreting cells
- β-cell precursors
- pancreatic islets
- transplantation
1. Introduction
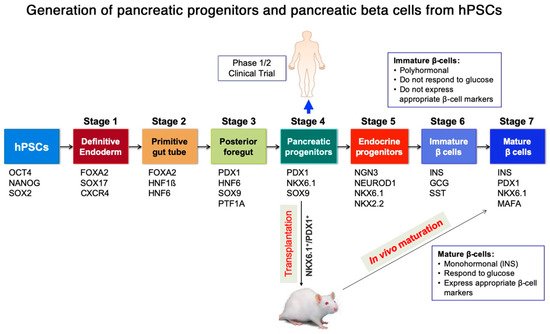
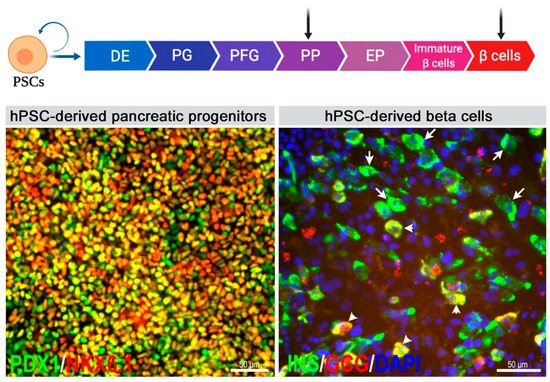
2. Generation of Pancreatic Progenitors and Beta Cells from Human Pluripotent Stem Cells (hPSCs)
| Feature | Pancreatic Progenitors | Pancreatic Beta Cells |
|---|---|---|
| Key transcription factors | PDX1, NKX6.1, and FOXA2 [7] | NKX6.1, MAFA, and PDX1 [7] |
| Surface markers | CD24 [32], CD142 [33], GP2 [22,34][22][34] | CD49a [35] |
| Duration of in vitro differentiation protocol | ~2 weeks [15,21][15][21] | ~30–36 days [17,18,19,36][17][18][19][36] |
| Method of differentiation | Monolayer [15,21][15][21] and aggregation in suspension [24,25][24][25] | Aggregation in suspension [17,18,19][17][18][19] |
| Display of human c-peptide secretion following transplantation in vivo (rodents) | ~3–4.5 months [9,22][9][22] | ~3–14 days [17,18,19][17][18][19] |
| After transplantation | Islet-like structure (INS+, GCG+, and SST+ cells) | INS+ cells |
| Clinical trials | Yes | No |
| HLA expression | Low [37] | High [37] |
| Off-target differentiation | Acinar and ductal cells could be co-generated [15,33][15][33] | Polyhormonal and other endocrine cells [8,17,19][8][17][19] |
| Generation of functionally relevant heterogenous beta cell subpopulations | undetermined | undetermined |
| Expansion and freeze-thaw potential for storage | Yes [38,39][38][39] | undetermined |
3. Intra-Islet Cell Contact and Communication for Functional Performance
Pancreatic islet architecture with the appropriate distribution of different endocrine cell types has been shown to contribute towards beta cell functionality and glucose responsiveness. Resonating from the findings that beta cells within an intact islet are more precise in their secretory and calcium response to elevated glucose levels than those in dissociated or dispersed islets [55[43][44][45],56,57], it is important to consider that intra-islet cell-cell communication between beta cells and other endocrine cells, as well as amongst different subpopulations of beta cells, is crucial for paracrine signaling and enhanced electrical coupling [60,61,62][46][47][48].3.1. Role of Pancreatic Endocrine Cells in Reversal of Hyperglycemia
3.2. Role of Heterogeneous Beta Cell Subtypes in Glucose Stimulated Insulin Secretion
4. Encapsulation of hPSC-Derived Pancreatic Progeny for Cell Therapy
The in vivo maturation of hPSC-derived pancreatic progenitors or pancreatic beta cells indispensably requires a suitable transplantation site, as well as an appropriate encapsulation material or device. The pancreas provides an appropriate microenvironment for the maturation of islets. However, a surgical method for delivery and retreivability has limited its consideration as a candidate for transplantation site. One study transplanted islets in rats and found that normoglycemia is achieved with fewer islets, compared to the extra-pancreatic sites, such as the liver and the kidney [92][78]. However, it is worth noting that pancreatic progenitors transplanted subcutaneously or under kidney capsules have, nevertheless, resulted in their differentiation into functional beta cells [15[15][30],30], despite not being exposed to a ‘pancreatic’ microenvironment. For example, a previous study investigated the effect of the transplantation site on the generation of monohormonal insulin-secreting cells and found that the transplantation of pancreatic progenitors under mammary fat pads or kidney capsules do not affect their maturation into beta cells [15]. It is plausible that the crucial vascular system supplying nutrients and oxygen to these transplanted progenitors carries cues sufficient to facilitate their maturation into beta cells. An encapsulation device is, therefore, a crucial factor affecting the performance of the transplanted cells and it can control teratoma formation if the transplanted cells are contaminated with undifferentiated cells. Encapsulation of the transplanted pancreatic lineage is crucial in case of T1D to prevent an autoimmune reaction against the transplanted cells. Additionally, if the cell therapy product is from an allogenic source, for example, commercialized off-the-shelf hPSC-derived beta cells, then encapsulation is still a requirement. However, in the case of T2D or monogenic diabetes, there is a possibility of transplanting pancreatic progeny derived from the patient’s own cells, that circumvents the need for encapsulation [5]. Achieving the right design of an encapsulation device requires putting together variables, such as the biocompatibility properties of the membrane, exposure to the blood stream, and availability of nutrition and oxygen for the encapsulated cells amongst others [93][79]. Studies are being done on modification of the available materials to improve these properties of the biomaterial, and have mainly been developed for islet transplantation, both in macro- and micro-encapsulation systems. The assessment of islet function and viability following their coating with alginate derivatives is being widely investigated for improving islet transplantation outcomes. Purified alginate improved the survival of encapsulated islets and had a moderate effect on necrosis compared to non-purified alginate capsules [94,95][80][81]. Furthermore, certain alginate modifications are particularly interesting to study as they could circumvent immune response following transplantation of allogenic islets. Modification of alginate capsules using triazole-derivatives showed positive results in preventing immune cell activation at capsule surfaces in mice and non-human primates [96][82]. The incorporation of the chemokine CXCL12 in the alginate capsule protected the islets and improved their function by serving as an immune-isolating material without the need for immune-suppression [97][83]. Likewise, such alginate-based microencapsulation methods are now being applied for stem cell therapy, such as for hPSC-derived beta cells. CXCL12 coating was recently shown to prolong the viability of hPSC-derived beta cells in immune-competent mice without requiring immunosuppression by preventing fibrotic overgrowth [98][84]. In addition, the CXCL12 coating enhanced beta cell function by improving their glucose responsiveness, thereby making it an important biomaterial to study further for beta cell encapsulation. While alginate-based microencapsulation is a promising option for beta cell replacement therapy, complete retrieval of the implanted microencapsulated islets in the portal vein, under kidney capsule or in the peritoneal space is challenging, often being invasive and incomplete [3,99,100][3][85][86]. Therefore, their use as an encapsulant for hPSC-derived beta cells, without a protective retrievable device could raise concerns as hPSC-derived beta cells have not yet been proven to be completely identical to pancreatic adult islets [27] and require their retrieval periodically to assess their viability, function, and to detect any teratomas formed. While studies on the formation of teratoma by unsorted terminally differentiated hPSC cultures containing target cells and uncommitted progenitors have revealed their expected frequency [101][87], it is unclear how the variability in differentiation efficiencies by different cell lines for the generation of beta cells would affect teratoma formation in vivo. However, the purification of fully mature hPSC-derived beta cells using surface markers prior to their microencapsulation could nullify some of these concerns. Therefore, further studies are required for the identification of these markers and their application. On the other hand, minimally invasive macro-encapsulation devices have also shown progress in delivering cell therapy products. The bilaminar TheraCyte macro-encapsulation device, which has an outer surface that promotes tissue engraftment or infiltration of blood vessels thus providing a close proximity of vasculature to the implanted cells and an inner membrane that prevents immune cell diffusion, has been shown to protect against immune-rejection of the transplanted pancreatic tissues in rodents. In addition, the device has allowed maturation of the hPSC-derived pancreatic lineages into pancreatic beta cells in animal models and proven competent at controlling hyperglycemia [9,29,31,102,103,104,105,106][9][29][31][88][89][90][91][92]. ViaCyte, leading the only clinical trial using hPSC-derived product for T1D treatment has reviewed the performance of its macro-encapsulation device, PEC-Encap, a TheraCyte adaptation, in the first phase of their study. The PEC-Encap is a combination of the hPSC-derived pancreatic progenitors enclosed in a semi-permeable, Encaptra drug delivery system, which was transplanted under the skin of T1D patients (https://viacyte.com/products/pec-encap-vc-01). The preliminary results indicated that the device is not effective in allowing its engraftment into the host tissue, which may impair supply of oxygen and nutrients to the therapeutic cells within the device (https://viacyte.com/products/pec-encap-vc-01). However, it was shown to protect the cells from immune invasion. To overcome these limitations, another ViaCyte system called the PEC-Direct, with a modified encapsulation membrane, is being currently employed in another clinical trial to assess its safety T1D hypoglycemic patients, however it requires immune-suppression (https://viacyte.com/products/pec-direct). Nonetheless, novel cell encapsulation systems are being developed, such as the Cell Pouch by Sernova (https://www.sernova.com/technology/) that facilitates formation of a pre-vascularized scaffold at the target site before cell delivery, that can advance cell therapy [107][93]. Another innovative device that aims to enhance oxygen availability to the encapsulated cells is the Beta-O2 device that consists of a gas chamber next to the encapsulated cells that allows the diffusion of oxygen to the cells (https://beta-o2.com/living-with-sair/). This chamber can be refilled occasionally to maintain a continuous supply of oxygen.References
- Cnop, M.; Welsh, N.; Jonas, J.C.; Jorns, A.; Lenzen, S.; Eizirik, D.L. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: Many differences, few similarities. Diabetes 2005, 54 (Suppl. 2), S97–S107.
- Murphy, R.; Ellard, S.; Hattersley, A.T. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat. Clin. Pract. Endocrinol. Metab. 2008, 4, 200–213.
- Vaithilingam, V.; Tuch, B.E. Islet transplantation and encapsulation: An update on recent developments. Rev. Diabet. Stud. 2011, 8, 51–67.
- Latres, E.; Finan, D.A.; Greenstein, J.L.; Kowalski, A.; Kieffer, T.J. Navigating Two Roads to Glucose Normalization in Diabetes: Automated Insulin Delivery Devices and Cell Therapy. Cell Metab. 2019, 29, 545–563.
- Abdelalim, E.M.; Bonnefond, A.; Bennaceur-Griscelli, A.; Froguel, P. Pluripotent stem cells as a potential tool for disease modelling and cell therapy in diabetes. Stem Cell Rev. Rep. 2014, 10, 327–337.
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872.
- Al-Khawaga, S.; Memon, B.; Butler, A.E.; Taheri, S.; Abou-Samra, A.B.; Abdelalim, E.M. Pathways governing development of stem cell-derived pancreatic beta cells: Lessons from embryogenesis. Biol. Rev. Camb. Philos. Soc. 2018, 93, 364–389.
- Rezania, A.; Bruin, J.E.; Arora, P.; Rubin, A.; Batushansky, I.; Asadi, A.; O’Dwyer, S.; Quiskamp, N.; Mojibian, M.; Albrecht, T.; et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 1121–1133.
- Rezania, A.; Bruin, J.E.; Xu, J.; Narayan, K.; Fox, J.K.; O’Neil, J.J.; Kieffer, T.J. Enrichment of human embryonic stem cell-derived NKX6.1-expressing pancreatic progenitor cells accelerates the maturation of insulin-secreting cells in vivo. Stem Cells 2013, 31, 2432–2442.
- Jennings, R.E.; Berry, A.A.; Kirkwood-Wilson, R.; Roberts, N.A.; Hearn, T.; Salisbury, R.J.; Blaylock, J.; Piper Hanley, K.; Hanley, N.A. Development of the human pancreas from foregut to endocrine commitment. Diabetes 2013, 62, 3514–3522.
- Taylor, B.L.; Liu, F.F.; Sander, M. Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Rep. 2013, 4, 1262–1275.
- Xu, X.; Browning, V.L.; Odorico, J.S. Activin, BMP and FGF pathways cooperate to promote endoderm and pancreatic lineage cell differentiation from human embryonic stem cells. Mech. Dev. 2011, 128, 412–427.
- Davenport, C.; Diekmann, U.; Budde, I.; Detering, N.; Naujok, O. Anterior-Posterior Patterning of Definitive Endoderm Generated from Human Embryonic Stem Cells Depends on the Differential Signaling of Retinoic Acid, Wnt-, and BMP-Signaling. Stem Cells 2016, 34, 2635–2647.
- Nostro, M.C.; Sarangi, F.; Ogawa, S.; Holtzinger, A.; Corneo, B.; Li, X.; Micallef, S.J.; Park, I.H.; Basford, C.; Wheeler, M.B.; et al. Stage-specific signaling through TGFbeta family members and WNT regulates patterning and pancreatic specification of human pluripotent stem cells. Development 2011, 138, 861–871.
- Nostro, M.C.; Sarangi, F.; Yang, C.; Holland, A.; Elefanty, A.G.; Stanley, E.G.; Greiner, D.L.; Keller, G. Efficient generation of NKX6-1+ pancreatic progenitors from multiple human pluripotent stem cell lines. Stem Cell Rep. 2015, 4, 591–604.
- Fujikura, J.; Hosoda, K.; Iwakura, H.; Tomita, T.; Noguchi, M.; Masuzaki, H.; Tanigaki, K.; Yabe, D.; Honjo, T.; Nakao, K. Notch/Rbp-j signaling prevents premature endocrine and ductal cell differentiation in the pancreas. Cell Metab. 2006, 3, 59–65.
- Russ, H.A.; Parent, A.V.; Ringler, J.J.; Hennings, T.G.; Nair, G.G.; Shveygert, M.; Guo, T.; Puri, S.; Haataja, L.; Cirulli, V.; et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015, 34, 1759–1772.
- Nair, G.G.; Liu, J.S.; Russ, H.A.; Tran, S.; Saxton, M.S.; Chen, R.; Juang, C.; Li, M.L.; Nguyen, V.Q.; Giacometti, S.; et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived β cells. Nat. Cell Biol. 2019, 21, 263–274.
- Pagliuca, F.W.; Millman, J.R.; Gurtler, M.; Segel, M.; Van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic beta cells in vitro. Cell 2014, 159, 428–439.
- Abdelalim, E.M.; Emara, M.M. Advances and challenges in the differentiation of pluripotent stem cells into pancreatic beta cells. World J. Stem Cells 2015, 7, 174–181.
- Memon, B.; Karam, M.; Al-Khawaga, S.; Abdelalim, E.M. Enhanced differentiation of human pluripotent stem cells into pancreatic progenitors co-expressing PDX1 and NKX6.1. Stem Cell Res. Ther. 2018, 9, 15.
- Cogger, K.F.; Sinha, A.; Sarangi, F.; McGaugh, E.C.; Saunders, D.; Dorrell, C.; Mejia-Guerrero, S.; Aghazadeh, Y.; Rourke, J.L.; Screaton, R.A.; et al. Glycoprotein 2 is a specific cell surface marker of human pancreatic progenitors. Nat. Commun. 2017, 8, 331.
- Aigha, I.I.; Memon, B.; Elsayed, A.K.; Abdelalim, E.M. Differentiation of human pluripotent stem cells into two distinct NKX6.1 populations of pancreatic progenitors. Stem Cell Res. Ther. 2018, 9, 83.
- Toyoda, T.; Mae, S.; Tanaka, H.; Kondo, Y.; Funato, M.; Hosokawa, Y.; Sudo, T.; Kawaguchi, Y.; Osafune, K. Cell aggregation optimizes the differentiation of human ESCs and iPSCs into pancreatic bud-like progenitor cells. Stem Cell Res. 2015, 14, 185–197.
- Jiang, W.; Shi, Y.; Zhao, D.; Chen, S.; Yong, J.; Zhang, J.; Qing, T.; Sun, X.; Zhang, P.; Ding, M.; et al. In vitro derivation of functional insulin-producing cells from human embryonic stem cells. Cell Res. 2007, 17, 333–344.
- D’Amour, K.A.; Bang, A.G.; Eliazer, S.; Kelly, O.G.; Agulnick, A.D.; Smart, N.G.; Moorman, M.A.; Kroon, E.; Carpenter, M.K.; Baetge, E.E. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat. Biotechnol. 2006, 24, 1392–1401.
- Hrvatin, S.; O’Donnell, C.W.; Deng, F.; Millman, J.R.; Pagliuca, F.W.; DiIorio, P.; Rezania, A.; Gifford, D.K.; Melton, D.A. Differentiated human stem cells resemble fetal, not adult, β cells. Proc. Natl. Acad. Sci. USA 2014, 111, 3038–3043.
- Bruin, J.E.; Erener, S.; Vela, J.; Hu, X.; Johnson, J.D.; Kurata, H.T.; Lynn, F.C.; Piret, J.M.; Asadi, A.; Rezania, A.; et al. Characterization of polyhormonal insulin-producing cells derived in vitro from human embryonic stem cells. Stem Cell Res. 2014, 12, 194–208.
- Agulnick, A.D.; Ambruzs, D.M.; Moorman, M.A.; Bhoumik, A.; Cesario, R.M.; Payne, J.K.; Kelly, J.R.; Haakmeester, C.; Srijemac, R.; Wilson, A.Z.; et al. Insulin-Producing Endocrine Cells Differentiated In Vitro From Human Embryonic Stem Cells Function in Macroencapsulation Devices In Vivo. Stem Cells Transl. Med. 2015, 4, 1214–1222.
- Rezania, A.; Bruin, J.E.; Riedel, M.J.; Mojibian, M.; Asadi, A.; Xu, J.; Gauvin, R.; Narayan, K.; Karanu, F.; O’Neil, J.J.; et al. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes 2012, 61, 2016–2029.
- Kroon, E.; Martinson, L.A.; Kadoya, K.; Bang, A.G.; Kelly, O.G.; Eliazer, S.; Young, H.; Richardson, M.; Smart, N.G.; Cunningham, J.; et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 2008, 26, 443–452.
- Jiang, W.; Sui, X.; Zhang, D.; Liu, M.; Ding, M.; Shi, Y.; Deng, H. CD24: A novel surface marker for PDX1-positive pancreatic progenitors derived from human embryonic stem cells. Stem Cells 2011, 29, 609–617.
- Kelly, O.G.; Chan, M.Y.; Martinson, L.A.; Kadoya, K.; Ostertag, T.M.; Ross, K.G.; Richardson, M.; Carpenter, M.K.; D’Amour, K.A.; Kroon, E.; et al. Cell-surface markers for the isolation of pancreatic cell types derived from human embryonic stem cells. Nat. Biotechnol. 2011, 29, 750–756.
- Ameri, J.; Borup, R.; Prawiro, C.; Ramond, C.; Schachter, K.A.; Scharfmann, R.; Semb, H. Efficient Generation of Glucose-Responsive Beta Cells from Isolated GP2. Cell Rep. 2017, 19, 36–49.
- Veres, A.; Faust, A.L.; Bushnell, H.L.; Engquist, E.N.; Kenty, J.H.; Harb, G.; Poh, Y.C.; Sintov, E.; Gürtler, M.; Pagliuca, F.W.; et al. Charting cellular identity during human in vitro β-cell differentiation. Nature 2019, 569, 368–373.
- Ramond, C.; Glaser, N.; Berthault, C.; Ameri, J.; Kirkegaard, J.S.; Hansson, M.; Honore, C.; Semb, H.; Scharfmann, R. Reconstructing human pancreatic differentiation by mapping specific cell populations during development. Elife 2017, 6, e27564.
- van der Torren, C.R.; Zaldumbide, A.; Duinkerken, G.; Brand-Schaaf, S.H.; Peakman, M.; Stange, G.; Martinson, L.; Kroon, E.; Brandon, E.P.; Pipeleers, D.; et al. Immunogenicity of human embryonic stem cell-derived beta cells. Diabetologia 2017, 60, 126–133.
- Trott, J.; Tan, E.K.; Ong, S.; Titmarsh, D.M.; Denil, S.; Giam, M.; Wong, C.K.; Wang, J.; Shboul, M.; Eio, M.; et al. Long-Term Culture of Self-renewing Pancreatic Progenitors Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2017, 8, 1675–1688.
- Konagaya, S.; Iwata, H. Chemically defined conditions for long-term maintenance of pancreatic progenitors derived from human induced pluripotent stem cells. Sci. Rep. 2019, 9, 640.
- Bocian-Sobkowska, J.; Zabel, M.; Wozniak, W.; Surdyk-Zasada, J. Polyhormonal aspect of the endocrine cells of the human fetal pancreas. Histochem. Cell Biol. 1999, 112, 147–153.
- Md Moin, A.S.; Dhawan, S.; Cory, M.; Butler, P.C.; Rizza, R.A.; Butler, A.E. Increased Frequency of Hormone Negative and Polyhormonal Endocrine Cells in Lean Individuals With Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 3628–3636.
- Riedel, M.J.; Asadi, A.; Wang, R.; Ao, Z.; Warnock, G.L.; Kieffer, T.J. Immunohistochemical characterisation of cells co-producing insulin and glucagon in the developing human pancreas. Diabetologia 2012, 55, 372–381.
- Scarl, R.T.; Corbin, K.L.; Vann, N.W.; Smith, H.M.; Satin, L.S.; Sherman, A.; Nunemaker, C.S. Intact pancreatic islets and dispersed beta-cells both generate intracellular calcium oscillations but differ in their responsiveness to glucose. Cell Calcium 2019, 83, 102081.
- Reissaus, C.A.; Piston, D.W. Reestablishment of Glucose Inhibition of Glucagon Secretion in Small Pseudoislets. Diabetes 2017, 66, 960–969.
- Benninger, R.K.; Head, W.S.; Zhang, M.; Satin, L.S.; Piston, D.W. Gap junctions and other mechanisms of cell-cell communication regulate basal insulin secretion in the pancreatic islet. J. Physiol. 2011, 589, 5453–5466.
- Johnston, N.R.; Mitchell, R.K.; Haythorne, E.; Pessoa, M.P.; Semplici, F.; Ferrer, J.; Piemonti, L.; Marchetti, P.; Bugliani, M.; Bosco, D.; et al. Beta Cell Hubs Dictate Pancreatic Islet Responses to Glucose. Cell Metab. 2016, 24, 389–401.
- Bavamian, S.; Klee, P.; Britan, A.; Populaire, C.; Caille, D.; Cancela, J.; Charollais, A.; Meda, P. Islet-cell-to-cell communication as basis for normal insulin secretion. Diabetes Obes Metab 2007, 9 (Suppl. 2), 118–132.
- Cabrera, O.; Berman, D.M.; Kenyon, N.S.; Ricordi, C.; Berggren, P.O.; Caicedo, A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc. Natl. Acad. Sci. USA 2006, 103, 2334–2339.
- Daunt, M.; Dale, O.; Smith, P.A. Somatostatin inhibits oxidative respiration in pancreatic beta-cells. Endocrinology 2006, 147, 1527–1535.
- Zhou, G.; Sinnett-Smith, J.; Liu, S.H.; Yu, J.; Wu, J.; Sanchez, R.; Pandol, S.J.; Abrol, R.; Nemunaitis, J.; Rozengurt, E.; et al. Down-regulation of pancreatic and duodenal homeobox-1 by somatostatin receptor subtype 5: A novel mechanism for inhibition of cellular proliferation and insulin secretion by somatostatin. Front. Physiol. 2014, 5, 226.
- Rodriguez-Diaz, R.; Dando, R.; Jacques-Silva, M.C.; Fachado, A.; Molina, J.; Abdulreda, M.H.; Ricordi, C.; Roper, S.D.; Berggren, P.O.; Caicedo, A. Alpha cells secrete acetylcholine as a non-neuronal paracrine signal priming beta cell function in humans. Nat. Med. 2011, 17, 888–892.
- Jain, R.; Lammert, E. Cell-cell interactions in the endocrine pancreas. Diabetes Obes. Metab. 2009, 11 (Suppl. 4), 159–167.
- Gromada, J.; Brock, B.; Schmitz, O.; Rorsman, P. Glucagon-like peptide-1: Regulation of insulin secretion and therapeutic potential. Basic Clin. Pharmacol. Toxicol. 2004, 95, 252–262.
- Fujita, Y.; Wideman, R.D.; Asadi, A.; Yang, G.K.; Baker, R.; Webber, T.; Zhang, T.; Wang, R.; Ao, Z.; Warnock, G.L.; et al. Glucose-dependent insulinotropic polypeptide is expressed in pancreatic islet alpha-cells and promotes insulin secretion. Gastroenterology 2010, 138, 1966–1975.
- van der Meulen, T.; Donaldson, C.J.; Caceres, E.; Hunter, A.E.; Cowing-Zitron, C.; Pound, L.D.; Adams, M.W.; Zembrzycki, A.; Grove, K.L.; Huising, M.O. Urocortin3 mediates somatostatin-dependent negative feedback control of insulin secretion. Nat. Med. 2015, 21, 769–776.
- Koh, D.S.; Cho, J.H.; Chen, L. Paracrine interactions within islets of Langerhans. J. Mol. Neurosci. 2012, 48, 429–440.
- Bansal, P.; Wang, Q. Insulin as a physiological modulator of glucagon secretion. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E751–E761.
- Henquin, J.C.; Rahier, J. Pancreatic alpha cell mass in European subjects with type 2 diabetes. Diabetologia 2011, 54, 1720–1725.
- Marroqui, L.; Masini, M.; Merino, B.; Grieco, F.A.; Millard, I.; Dubois, C.; Quesada, I.; Marchetti, P.; Cnop, M.; Eizirik, D.L. Pancreatic alpha Cells are Resistant to Metabolic Stress-induced Apoptosis in Type 2 Diabetes. EBioMedicine 2015, 2, 378–385.
- Bonnet-Serrano, F.; Diedisheim, M.; Mallone, R.; Larger, E. Decreased α-cell mass and early structural alterations of the exocrine pancreas in patients with type 1 diabetes: An analysis based on the nPOD repository. PLoS ONE 2018, 13, e0191528.
- Tomei, A.A.; Villa, C.; Ricordi, C. Development of an encapsulated stem cell-based therapy for diabetes. Expert. Opin. Biol. Ther. 2015, 15, 1321–1336.
- Fraker, C.A.; Ricordi, C.; Inverardi, L.; Domínguez-Bendala, J. Oxygen: A master regulator of pancreatic development? Biol. Cell 2009, 101, 431–440.
- Shih, H.P.; Kopp, J.L.; Sandhu, M.; Dubois, C.L.; Seymour, P.A.; Grapin-Botton, A.; Sander, M. A Notch-dependent molecular circuitry initiates pancreatic endocrine and ductal cell differentiation. Development 2012, 139, 2488–2499.
- Merani, S.; Toso, C.; Emamaullee, J.; Shapiro, A.M. Optimal implantation site for pancreatic islet transplantation. Br. J. Surg. 2008, 95, 1449–1461.
- Roscioni, S.S.; Migliorini, A.; Gegg, M.; Lickert, H. Impact of islet architecture on beta-cell heterogeneity, plasticity and function. Nat. Rev. Endocrinol. 2016, 12, 695–709.
- Heimberg, H.; De Vos, A.; Vandercammen, A.; Van Schaftingen, E.; Pipeleers, D.; Schuit, F. Heterogeneity in glucose sensitivity among pancreatic beta-cells is correlated to differences in glucose phosphorylation rather than glucose transport. EMBO J. 1993, 12, 2873–2879.
- Pipeleers, D.; Kiekens, R.; Ling, Z.; Wilikens, A.; Schuit, F. Physiologic relevance of heterogeneity in the pancreatic beta-cell population. Diabetologia 1994, 37 (Suppl. 2), S57–s64.
- Pipeleers, D.; De Mesmaeker, I.; Robert, T.; Van Hulle, F. Heterogeneity in the Beta-Cell Population: A Guided Search Into Its Significance in Pancreas and in Implants. Curr. Diab. Rep. 2017, 17, 86.
- Wojtusciszyn, A.; Armanet, M.; Morel, P.; Berney, T.; Bosco, D. Insulin secretion from human beta cells is heterogeneous and dependent on cell-to-cell contacts. Diabetologia 2008, 51, 1843–1852.
- Westacott, M.J.; Ludin, N.W.F.; Benninger, R.K.P. Spatially Organized β-Cell Subpopulations Control Electrical Dynamics across Islets of Langerhans. Biophys. J. 2017, 113, 1093–1108.
- Bader, E.; Migliorini, A.; Gegg, M.; Moruzzi, N.; Gerdes, J.; Roscioni, S.S.; Bakhti, M.; Brandl, E.; Irmler, M.; Beckers, J.; et al. Identification of proliferative and mature β-cells in the islets of Langerhans. Nature 2016, 535, 430–434.
- Meda, P. Protein-mediated interactions of pancreatic islet cells. Scientifica (Cairo) 2013, 2013, 621249.
- Speier, S.; Gjinovci, A.; Charollais, A.; Meda, P.; Rupnik, M. Cx36-mediated coupling reduces beta-cell heterogeneity, confines the stimulating glucose concentration range, and affects insulin release kinetics. Diabetes 2007, 56, 1078–1086.
- Ravier, M.A.; Güldenagel, M.; Charollais, A.; Gjinovci, A.; Caille, D.; Söhl, G.; Wollheim, C.B.; Willecke, K.; Henquin, J.C.; Meda, P. Loss of connexin36 channels alters beta-cell coupling, islet synchronization of glucose-induced Ca2+ and insulin oscillations, and basal insulin release. Diabetes 2005, 54, 1798–1807.
- Konstantinova, I.; Nikolova, G.; Ohara-Imaizumi, M.; Meda, P.; Kucera, T.; Zarbalis, K.; Wurst, W.; Nagamatsu, S.; Lammert, E. EphA-Ephrin-A-mediated beta cell communication regulates insulin secretion from pancreatic islets. Cell 2007, 129, 359–370.
- Do Hoang, O.; Thorn, P. Insulin secretion from beta cells within intact islets: Location matters. Clin. Exp. Pharmacol. Physiol. 2015, 42, 406–414.
- Efrat, S. Beta-Cell Dedifferentiation in Type 2 Diabetes: Concise Review. Stem Cells 2019, 37, 1267–1272.
- Stagner, J.I.; Rilo, H.L.; White, K.K. The pancreas as an islet transplantation site. Confirmation in a syngeneic rodent and canine autotransplant model. JOP 2007, 8, 628–636.
- Opara, E.C.; Mirmalek-Sani, S.H.; Khanna, O.; Moya, M.L.; Brey, E.M. Design of a bioartificial pancreas(+). J. Investig. Med. 2010, 58, 831–837.
- Mallett, A.G.; Korbutt, G.S. Alginate modification improves long-term survival and function of transplanted encapsulated islets. Tissue Eng. Part A 2009, 15, 1301–1309.
- Langlois, G.; Dusseault, J.; Bilodeau, S.; Tam, S.K.; Magassouba, D.; Hallé, J.P. Direct effect of alginate purification on the survival of islets immobilized in alginate-based microcapsules. Acta Biomater. 2009, 5, 3433–3440.
- Vegas, A.J.; Veiseh, O.; Doloff, J.C.; Ma, M.; Tam, H.H.; Bratlie, K.; Li, J.; Bader, A.R.; Langan, E.; Olejnik, K.; et al. Combinatorial hydrogel library enables identification of materials that mitigate the foreign body response in primates. Nat. Biotechnol. 2016, 34, 345–352.
- Chen, T.; Yuan, J.; Duncanson, S.; Hibert, M.L.; Kodish, B.C.; Mylavaganam, G.; Maker, M.; Li, H.; Sremac, M.; Santosuosso, M.; et al. Alginate encapsulant incorporating CXCL12 supports long-term allo- and xenoislet transplantation without systemic immune suppression. Am. J. Transplant. 2015, 15, 618–627.
- Alagpulinsa, D.A.; Cao, J.J.L.; Driscoll, R.K.; Sîrbulescu, R.F.; Penson, M.F.E.; Sremac, M.; Engquist, E.N.; Brauns, T.A.; Markmann, J.F.; Melton, D.A.; et al. Alginate-microencapsulation of human stem cell-derived β cells with CXCL12 prolongs their survival and function in immunocompetent mice without systemic immunosuppression. Am. J. Transplant. 2019, 19, 1930–1940.
- Tuch, B.E.; Keogh, G.W.; Williams, L.J.; Wu, W.; Foster, J.L.; Vaithilingam, V.; Philips, R. Safety and viability of microencapsulated human islets transplanted into diabetic humans. Diabetes Care 2009, 32, 1887–1889.
- Paredes-Juarez, G.A.; de Vos, P.; Bulte, J.W.M. Recent progress in the use and tracking of transplanted islets as a personalized treatment for type 1 diabetes. Expert Rev. Precis. Med. Drug Dev. 2017, 2, 57–67.
- Hentze, H.; Soong, P.L.; Wang, S.T.; Phillips, B.W.; Putti, T.C.; Dunn, N.R. Teratoma formation by human embryonic stem cells: Evaluation of essential parameters for future safety studies. Stem Cell Res. 2009, 2, 198–210.
- Kirk, K.; Hao, E.; Lahmy, R.; Itkin-Ansari, P. Human embryonic stem cell derived islet progenitors mature inside an encapsulation device without evidence of increased biomass or cell escape. Stem Cell Res. 2014, 12, 807–814.
- Yakhnenko, I.; Wong, W.K.; Katkov, I.I.; Itkin-Ansari, P. Cryopreservation of human insulin expressing cells macro-encapsulated in a durable therapeutic immunoisolating device theracyte. Cryo. Lett. 2012, 33, 518–531.
- Robert, T.; De Mesmaeker, I.; Stangé, G.M.; Suenens, K.G.; Ling, Z.; Kroon, E.J.; Pipeleers, D.G. Functional Beta Cell Mass from Device-Encapsulated hESC-Derived Pancreatic Endoderm Achieving Metabolic Control. Stem Cell Rep. 2018, 10, 739–750.
- Bruin, J.E.; Rezania, A.; Xu, J.; Narayan, K.; Fox, J.K.; O’Neil, J.J.; Kieffer, T.J. Maturation and function of human embryonic stem cell-derived pancreatic progenitors in macroencapsulation devices following transplant into mice. Diabetologia 2013, 56, 1987–1998.
- Lee, S.H.; Hao, E.; Savinov, A.Y.; Geron, I.; Strongin, A.Y.; Itkin-Ansari, P. Human beta-cell precursors mature into functional insulin-producing cells in an immunoisolation device: Implications for diabetes cell therapies. Transplantation 2009, 87, 983–991.
- Schweicher, J.; Nyitray, C.; Desai, T.A. Membranes to achieve immunoprotection of transplanted islets. Front. Biosci. 2014, 19, 49–76.