Carbohydrate is the most abundant macronutrient in the human diet, providing 45–70% of daily calories.
- insulin resistance
- macronutrients
- obesity
1. Carbohydrate
Carbohydrate is the most abundant macronutrient in the human diet, providing 45–70% of daily calories [3,4]. However, carbohydrates are not considered an essential nutrient for humans [17], and their increased consumption has recently been associated with “carbotoxicity” [6,7]. Several human trials have shown that reducing carbohydrate consumption is beneficial for metabolic health, and ketogenic diets that severely limit carbohydrate intake to <10% daily energy are effective in producing weight loss and improving the glycaemic profile in type 2 diabetes [18]. Furthermore, a recent large-scale epidemiological study (Prospective Urban Rural Epidemiology (PURE) study) showed that a high carbohydrate intake (highest (quintile 5) vs. lowest quintile (quintile 1)) is associated with increased risk of mortality and dyslipidaemia [7,19]. Together, these observations make a case for revising current dietary guidelines to reduce the total amount of carbohydrate consumption and decrease the proportion of daily energy derived from carbohydrate.
Carbohydrate is the most abundant macronutrient in the human diet, providing 45–70% of daily calories [1][2]. However, carbohydrates are not considered an essential nutrient for humans [3], and their increased consumption has recently been associated with “carbotoxicity” [4][5]. Several human trials have shown that reducing carbohydrate consumption is beneficial for metabolic health, and ketogenic diets that severely limit carbohydrate intake to <10% daily energy are effective in producing weight loss and improving the glycaemic profile in type 2 diabetes [6]. Furthermore, a recent large-scale epidemiological study (Prospective Urban Rural Epidemiology (PURE) study) showed that a high carbohydrate intake (highest (quintile 5) vs. lowest quintile (quintile 1)) is associated with increased risk of mortality and dyslipidaemia [5][7]. Together, these observations make a case for revising current dietary guidelines to reduce the total amount of carbohydrate consumption and decrease the proportion of daily energy derived from carbohydrate.
2. Types of Carbohydrates and Their Metabolic Effects
In addition to total carbohydrate intake, evidence suggests that the “type” of carbohydrate eaten is also an important determinant of metabolic outcomes [8][9][10][11][12]. Fibre, starch, sucrose, and high fructose corn syrup (HFCS) are the major types of carbohydrates in the diet of adult humans [8][9][10][11][12]. Fibre is not a major source of daily energy [2][13]. It is composed of polysaccharides derived from the plant cell wall, whole grains, fruits, and vegetables, and includes resistant starch, inulin, oligofructose, polydextrose, and galactooligosaccharides [9]. Studies have shown that consuming fibre-rich foods leads to metabolic improvements such as weight loss and improved insulin sensitivity, and these changes are associated with an increased abundance of beneficial bacteria in the gut microbiome [9][10][14]. Currently, the daily intake of fibre for the U.S. population is 16 g/d; for European adults, the average intake is 20–25 g/d, which is less than the recommended fibre intake of 25-38 g/d by the U.S. Department of Agriculture (USDA) [13][15].
In terms of energy, starch, sucrose, and HFCS account for most of the carbohydrate-derived calories in adult Western diets [2]. Starch is a polysaccharide of glucose that exists as either linear chains of glucose monomers joined to each other by α-1,4 glycosidic bonds (amylose) or in the branched form containing both α-1,6 and α-1,4 linkages (amylopectin) [16]. Sucrose (table sugar) is a disaccharide of glucose and fructose, while HFCS is a mixture of glucose and fructose in monosaccharide form [2][17]. HFCS is produced by treating corn syrup with the enzyme “glucose isomerase” that converts glucose derived from corn starch to fructose [12]. In the U.S., successful glucose isomerisation on an industrial scale in 1967 led to the replacement of sucrose by HFCS in processed foods because of greater availability and lower prices of corn [12][18]. While sucrose is still the predominant caloric sweetener in most parts of the world, HFCS accounts for ~40% of caloric sweeteners added to foods (e.g., canned fruits, jellies, and baked goods etc) and drinks in the U.S. [12][19][20]. The most commonly used forms of HFCS are HFCS-55 (containing 55% fructose and 45% glucose) and HFCS-42 [19][20].
Glucose and fructose are the monomeric building blocks of the major energy-providing dietary carbohydrates [2]. Glucose is a source of energy for all tissues, but fructose is not essential for human metabolism and it is rarely consumed in isolation [11][19]. Fructose can be synthesised endogenously in the liver from glucose via the polyol pathway [21]. In general, most Western diets contain over three-fold more glucose than fructose [22]. In contrast to fructose found in nutrient-rich whole fruits, consuming fructose-derived calories from nutrient-poor sources such as caloric sweeteners (sucrose, HFCS and pure fructose) added to processed foods and beverages have been associated with adverse metabolic consequences [11][12]. For example, epidemiological data from the U.S. suggests that increased consumption of fructose, especially from HFCS, is responsible for the rapid rise in obesity prevalence over the last 40–50 years [12][23]. The increase in fructose and HFCS consumption closely paralleled the rise in prevalence of overweight and obesity in the U.S. between 1960 and 2000 [12][23]. In addition to obesity, higher intakes of sucrose, HFCS, and fructose, especially from beverages, increase the risk of developing type 2 diabetes [24][25][26][27]. Similarly, experimental studies in animals and humans have shown that consuming extra calories from fructose-containing sweeteners promote obesity, dyslipidaemia, fatty liver, and insulin resistance [11][28][29][30][31][32][33][34]. These adverse effects are more obvious when fructose is derived from beverages such as soda drinks, sweetened milk drinks, fruit juices, and iced tea [33][35][36][37]. This is because, compared with solid foods, the biological regulation of total energy intake is less precise when sugars are consumed as fluids [2][38][39]. Furthermore, fructose intake stimulates parts of the brain associated with feelings of reward and pleasure, and these hedonic effects of fructose can promote increased energy consumption [40]. Functional MRI scanning in healthy volunteers after a glucose or fructose drink showed that fructose stimulated greater reactivity to food cues in the visual cortex and left orbital frontal cortex [41]. Behavioural studies in animals have also shown signs of dependence, such as bingeing, withdrawal, craving, and cross-sensitisation to other drugs with intermittent sucrose administration [42]. However, contrary to the effects of consuming excess calories from fructose, results of the studies comparing the metabolic consequences of fructose-containing caloric sweeteners with other types of carbohydrates (e.g., glucose, maltose and starch) in energy-matched experiments have been inconsistent and contradictory [31][33][43][44]. This has led to strong controversy in sugar research and has caused debate as to whether fructose per se is harmful to health beyond its contribution to excess calories [33][43].
3. Glycaemic Index and Metabolic Effects of Carbohydrates
In addition to molecular structure, the glycaemic index (GI) is also used as a marker of carbohydrate quality. GI is a measure of the glycaemic response to consuming a food item containing 50 g of carbohydrate. It is measured as the incremental AUC (iAUC) for blood glucose over a two-hour timeframe and expressed as a percentage of the iAUC of a reference food, usually, pure glucose solution or white bread, which are assigned GI values of 100 (GI = (iAUC
food item
/iAUC
reference) × 100) [57,58]. A related parameter is the glycaemic load (GL), which is the product of GI and available carbohydrate in a given amount of food (GL = GI × available carbohydrate in the food item) [57]. Foods with GI values of ≤55 are classified as low GI foods; those with GI of 56–69 are medium; and those with GI ≥ 70 are labelled as high GI foods [57]. Most starchy foods have a GI of >70, and Thai Jasmine rice has a GI of 100 [59]. Compared with low GI diets, consuming high GI diets would produce a greater spike in postprandial blood glucose and insulin concentrations [58]. This will result in the rapid utilisation of glucose by peripheral tissues, leading to a faster return of feelings of hunger, and the spike of insulin could lead to greater anabolic effects of insulin such as lipogenesis and increased lipidaemia. This could lead to adverse consequences such as obesity and insulin resistance in the long term [58]. In addition, emerging evidence suggests that high GI diets could have transgenerational effects due to epigenetic changes induced in the placental and foetal tissues [60,61].
) × 100) [45][46]. A related parameter is the glycaemic load (GL), which is the product of GI and available carbohydrate in a given amount of food (GL = GI × available carbohydrate in the food item) [45]. Foods with GI values of ≤55 are classified as low GI foods; those with GI of 56–69 are medium; and those with GI ≥ 70 are labelled as high GI foods [45]. Most starchy foods have a GI of >70, and Thai Jasmine rice has a GI of 100 [47]. Compared with low GI diets, consuming high GI diets would produce a greater spike in postprandial blood glucose and insulin concentrations [46]. This will result in the rapid utilisation of glucose by peripheral tissues, leading to a faster return of feelings of hunger, and the spike of insulin could lead to greater anabolic effects of insulin such as lipogenesis and increased lipidaemia. This could lead to adverse consequences such as obesity and insulin resistance in the long term [46]. In addition, emerging evidence suggests that high GI diets could have transgenerational effects due to epigenetic changes induced in the placental and foetal tissues [48][49].
Results of experimental studies comparing the effects of low vs. high GI diets on subjective measures of satiety, fullness and appetite have been inconsistent [57]. Although some studies have reported higher ratings of fullness in subjects consuming low GI diets [62,63], others have reported no significant differences in satiety on low vs. high GI foods [64,65,66]. Furthermore, data from studies investigating the relationship between dietary GI and obesity have been equivocal [57]. A cross-sectional study in young Japanese women showed a positive correlation between GI, GL, and body mass index (BMI) [67]. Similarly, in a study involving 175 subjects with type 2 diabetes, there was a positive association between dietary GI and waist circumference [68]. In an eight-week weight-loss trial including 30% energy restriction, subjects on a low GI diet lost significantly more weight than those in the high GI group [69]. At a molecular level, a study in male subjects given a high or an isocaloric low GI meal after exercise, showed that glucose and insulin AUC were increased, while gene expression of fatty acid transporter CD36 in skeletal muscle tissue was significantly reduced after consuming a high GI meal [70]. This indicates reduced fat metabolism after consuming a high GI meal [70]. Contrary to these observations, no association between dietary GI and BMI was observed in a study of Spanish adults [71]. Moreover, in older adults from the United Kingdom, no association was reported between GI and body weight or BMI [72]. This was similar to the data for elderly subjects from rural Spain, showing no association between GI or GL and waist circumference or BMI [73]. The results of these epidemiological studies are consistent with several weight-loss trials, including those that involved energy-matched interventions, which reported no differences in weight loss in subjects maintained on high vs. low GI diets [74,75,76,77]. The evidence from the prospective studies for the association between dietary GI and risk of developing type 2 diabetes [78,79,80,81], and results of experimental trials exploring the link between GI and markers of glycaemic control, have also yielded contradictory evidence [75,82,83].
Results of experimental studies comparing the effects of low vs. high GI diets on subjective measures of satiety, fullness and appetite have been inconsistent [45]. Although some studies have reported higher ratings of fullness in subjects consuming low GI diets [50][51], others have reported no significant differences in satiety on low vs. high GI foods [52][53][54]. Furthermore, data from studies investigating the relationship between dietary GI and obesity have been equivocal [45]. A cross-sectional study in young Japanese women showed a positive correlation between GI, GL, and body mass index (BMI) [55]. Similarly, in a study involving 175 subjects with type 2 diabetes, there was a positive association between dietary GI and waist circumference [56]. In an eight-week weight-loss trial including 30% energy restriction, subjects on a low GI diet lost significantly more weight than those in the high GI group [57]. At a molecular level, a study in male subjects given a high or an isocaloric low GI meal after exercise, showed that glucose and insulin AUC were increased, while gene expression of fatty acid transporter CD36 in skeletal muscle tissue was significantly reduced after consuming a high GI meal [58]. This indicates reduced fat metabolism after consuming a high GI meal [58]. Contrary to these observations, no association between dietary GI and BMI was observed in a study of Spanish adults [59]. Moreover, in older adults from the United Kingdom, no association was reported between GI and body weight or BMI [60]. This was similar to the data for elderly subjects from rural Spain, showing no association between GI or GL and waist circumference or BMI [61]. The results of these epidemiological studies are consistent with several weight-loss trials, including those that involved energy-matched interventions, which reported no differences in weight loss in subjects maintained on high vs. low GI diets [62][63][64][65]. The evidence from the prospective studies for the association between dietary GI and risk of developing type 2 diabetes [66][67][68][69], and results of experimental trials exploring the link between GI and markers of glycaemic control, have also yielded contradictory evidence [63][70][71].
Possible reasons for the inconsistent evidence about the metabolic outcomes associated with high vs. low GI diets include confounding factors such as the higher fibre content of low GI diets and the use of food frequency questionnaires in observational studies for collecting self-reported dietary data [57]. Another issue with the concept of GI is that foods containing sucrose or HFCS can have low GI values (e.g., the GI of pure fructose is 19) but still be adverse for metabolic health [22,84]. However, a recent meta-analysis of prospective cohort studies involving healthy adults showed a 90% increase in the risk of type 2 diabetes when comparing the lowest to the highest GI exposure across the globe (GI of 48 vs. 76) [85]. The strong association with high dietary GI and risk of type 2 diabetes was independent of levels of dietary fibre intake [85]. Furthermore, a recent publication from the high-profile PURE study involving data for 137,851 subjects from 20 countries on five continents showed an increased risk of cardiovascular disease and death with the high GI of the diet [86]. When comparing the highest vs. lowest quantiles of GI, the risk of a composite outcome of major cardiovascular event and death was increased both in subjects with (hazard ratio 1.51) and without (hazard ratio 1.21) pre-existing cardiovascular disease [86]. The WHO commissioned a systematic review and meta-analysis of prospective studies and randomised clinical trials to investigate the association between the intake of dietary fibre, whole grains, GI, and cardiometabolic disease [22]. When compared with people that consumed low fibre diets, coronary artery disease, type 2 diabetes and all-cause mortality were decreased by 15–30% in high fibre consumers [22]. The observational data for mortality translated into 13 fewer deaths for highest vs. lowest fibre intake per 1000 participants over the course of the studies [22]. In experimental trials, high fibre intake resulted in benefits such as lower body weight and lower levels of cholesterol [22]. The results of whole-grain consumption were similar to fibre intake, but a reduction in the risk of type 2 diabetes with the low vs. high GI diet was modest when compared with data for fibre intake [22]. Similarly, data from clinical trials showed inconsistent effects of GI on cardiometabolic outcomes [22]. Thus, overall, the evidence for using low GI diets as a strategy for the prevention and treatment of cardiometabolic disease is not as strong as that for high fibre intake [22,57]. Further well-controlled studies are required to formulate dietary guidelines in the light of the impact of GI on cardiometabolic outcomes.
Possible reasons for the inconsistent evidence about the metabolic outcomes associated with high vs. low GI diets include confounding factors such as the higher fibre content of low GI diets and the use of food frequency questionnaires in observational studies for collecting self-reported dietary data [45]. Another issue with the concept of GI is that foods containing sucrose or HFCS can have low GI values (e.g., the GI of pure fructose is 19) but still be adverse for metabolic health [10][72]. However, a recent meta-analysis of prospective cohort studies involving healthy adults showed a 90% increase in the risk of type 2 diabetes when comparing the lowest to the highest GI exposure across the globe (GI of 48 vs. 76) [73]. The strong association with high dietary GI and risk of type 2 diabetes was independent of levels of dietary fibre intake [73]. Furthermore, a recent publication from the high-profile PURE study involving data for 137,851 subjects from 20 countries on five continents showed an increased risk of cardiovascular disease and death with the high GI of the diet [74]. When comparing the highest vs. lowest quantiles of GI, the risk of a composite outcome of major cardiovascular event and death was increased both in subjects with (hazard ratio 1.51) and without (hazard ratio 1.21) pre-existing cardiovascular disease [74]. The WHO commissioned a systematic review and meta-analysis of prospective studies and randomised clinical trials to investigate the association between the intake of dietary fibre, whole grains, GI, and cardiometabolic disease [10]. When compared with people that consumed low fibre diets, coronary artery disease, type 2 diabetes and all-cause mortality were decreased by 15–30% in high fibre consumers [10]. The observational data for mortality translated into 13 fewer deaths for highest vs. lowest fibre intake per 1000 participants over the course of the studies [10]. In experimental trials, high fibre intake resulted in benefits such as lower body weight and lower levels of cholesterol [10]. The results of whole-grain consumption were similar to fibre intake, but a reduction in the risk of type 2 diabetes with the low vs. high GI diet was modest when compared with data for fibre intake [10]. Similarly, data from clinical trials showed inconsistent effects of GI on cardiometabolic outcomes [10]. Thus, overall, the evidence for using low GI diets as a strategy for the prevention and treatment of cardiometabolic disease is not as strong as that for high fibre intake [10][45]. Further well-controlled studies are required to formulate dietary guidelines in the light of the impact of GI on cardiometabolic outcomes.
4. Molecular Mechanisms of Metabolic Benefits of Fibre Intake
The mechanisms of beneficial effects of dietary fibre on gut microbiota composition and function as well as on host metabolism are well established [21]. Bacteria in the cecum and colon have the enzymatic machinery to ferment dietary fibre into short-chain fatty acids (SCFAs), mainly acetate, butyrate, and propionate at a ratio 3:1:1 which are absorbed into the systemic circulation. The binding of these fatty acids to G-protein-coupled receptors (GPCRs) in various tissues is thought to mediate the metabolic effects of fibre intake [21,25,87,88]. Butyrate, acetate, and propionate bind, with the highest selectivity, to the G-protein coupled receptors GPR109A, GPR43 and GPR41, respectively [87,88,89]. Butyrate is the major source of energy for enterocytes in the gut, while propionate and acetate might be metabolised in the liver. Acetate has the most marked systemic effects with plasma concentrations reaching 19–160 µM vs. 1–13 µM for butyrate and propionate [21].
The mechanisms of beneficial effects of dietary fibre on gut microbiota composition and function as well as on host metabolism are well established [9]. Bacteria in the cecum and colon have the enzymatic machinery to ferment dietary fibre into short-chain fatty acids (SCFAs), mainly acetate, butyrate, and propionate at a ratio 3:1:1 which are absorbed into the systemic circulation. The binding of these fatty acids to G-protein-coupled receptors (GPCRs) in various tissues is thought to mediate the metabolic effects of fibre intake [9][13][75][76]. Butyrate, acetate, and propionate bind, with the highest selectivity, to the G-protein coupled receptors GPR109A, GPR43 and GPR41, respectively [75][76][77]. Butyrate is the major source of energy for enterocytes in the gut, while propionate and acetate might be metabolised in the liver. Acetate has the most marked systemic effects with plasma concentrations reaching 19–160 µM vs. 1–13 µM for butyrate and propionate [9].
Resistant starch, made of linear amylose chains in granules that are resistant to digestion by intestinal enzymes, is one of the most commonly used types of fibre in research. Mice fed on resistant starch had increased glycolysis and fatty acid oxidation in the liver [90]. It is also known to beneficially reshape their gut microbiota by increasing the abundance of Bacteroides, Akkermansia, and Bifidobacterium while reducing Firmicutes [90]. Similarly, in the setting of isocaloric diets, replacing 30% energy from starch with resistant starch for 12 weeks produced an increase in the concentration of all three SCFAs in the cecum, reduced body weight and adiposity by increasing energy expenditure and oxidative lipid metabolism, and increased insulin sensitivity in mice [21]. Compared with native starch-fed mice, mice consuming a high resistant starch diet had drastically different plasma metabolome, including a 22-fold increase in the circulating concentrations of the tryptophan-derived metabolite indole propionate [91]. Treatment of rats with indole propionate resulted in an improved glycaemic profile [92]. In humans, the consumption of resistant starch lowered cholesterol and body fat, substantially increased acetate and propionate concentrations in plasma, and improved insulin sensitivity in subjects with metabolic syndrome [89,93].
Resistant starch, made of linear amylose chains in granules that are resistant to digestion by intestinal enzymes, is one of the most commonly used types of fibre in research. Mice fed on resistant starch had increased glycolysis and fatty acid oxidation in the liver [78]. It is also known to beneficially reshape their gut microbiota by increasing the abundance of Bacteroides, Akkermansia, and Bifidobacterium while reducing Firmicutes [78]. Similarly, in the setting of isocaloric diets, replacing 30% energy from starch with resistant starch for 12 weeks produced an increase in the concentration of all three SCFAs in the cecum, reduced body weight and adiposity by increasing energy expenditure and oxidative lipid metabolism, and increased insulin sensitivity in mice [9]. Compared with native starch-fed mice, mice consuming a high resistant starch diet had drastically different plasma metabolome, including a 22-fold increase in the circulating concentrations of the tryptophan-derived metabolite indole propionate [79]. Treatment of rats with indole propionate resulted in an improved glycaemic profile [80]. In humans, the consumption of resistant starch lowered cholesterol and body fat, substantially increased acetate and propionate concentrations in plasma, and improved insulin sensitivity in subjects with metabolic syndrome [77][81].
Intake of SCFAs reproduces most of the benefits associated with increased fibre intake. Most of the mouse studies have focused on the high-fat diet (HFD) model of obesity, and diets containing 5% (wt./wt.) of individual SCFAs have been commonly used. Supplementation of HFD diets with SCFAs either completely or partially prevented HFD-induced obesity without any changes in physical activity [94]. Acetate was found to be most effective of all the SCFAs in reducing body weight in some studies, while others showed an equal effect for all three SCFAs [94]. In addition, a 12-week HFD mouse study showed an increased energy expenditure and fatty acid oxidation secondary to increased AMPK activity and UCP2 expression in the liver with SCFAs supplementation [95]. This led to about a 1.5-fold higher glucose infusion rate in animals fed any of the three SCFA-containing diets during hyperinsulinemic clamp studies, indicating improved insulin sensitivity [95]. Improvement in glucose tolerance, lower fasting insulin and blood glucose, mitochondrial biogenesis, beige adipogenesis, and increased UCP-1 expression in brown fat have also been reported with SCFA intake [96,97,98]. Similar to resistant starch consumption, a decrease in the proportion of Firmicutes and an increase in Bacteroides was observed with SCFA intake [94]. In obese humans, consumption of 1.5 g/day acetate for 12 weeks reduced body weight and BMI and caused a 3.5% decrease in the abdominal fat area [99].
Intake of SCFAs reproduces most of the benefits associated with increased fibre intake. Most of the mouse studies have focused on the high-fat diet (HFD) model of obesity, and diets containing 5% (wt./wt.) of individual SCFAs have been commonly used. Supplementation of HFD diets with SCFAs either completely or partially prevented HFD-induced obesity without any changes in physical activity [82]. Acetate was found to be most effective of all the SCFAs in reducing body weight in some studies, while others showed an equal effect for all three SCFAs [82]. In addition, a 12-week HFD mouse study showed an increased energy expenditure and fatty acid oxidation secondary to increased AMPK activity and UCP2 expression in the liver with SCFAs supplementation [83]. This led to about a 1.5-fold higher glucose infusion rate in animals fed any of the three SCFA-containing diets during hyperinsulinemic clamp studies, indicating improved insulin sensitivity [83]. Improvement in glucose tolerance, lower fasting insulin and blood glucose, mitochondrial biogenesis, beige adipogenesis, and increased UCP-1 expression in brown fat have also been reported with SCFA intake [84][85][86]. Similar to resistant starch consumption, a decrease in the proportion of Firmicutes and an increase in Bacteroides was observed with SCFA intake [82]. In obese humans, consumption of 1.5 g/day acetate for 12 weeks reduced body weight and BMI and caused a 3.5% decrease in the abdominal fat area [87].
GPCR activity is required for the metabolic benefits of SCFAs. HFD feeding reduces the expression of GPR-41, 43 and 109A in the adipose tissue of animals [97,100]. Studies using knockout mice have been critical to understanding the mechanisms of the metabolic effects of SCFAs. GPR41
GPCR activity is required for the metabolic benefits of SCFAs. HFD feeding reduces the expression of GPR-41, 43 and 109A in the adipose tissue of animals [85][88]. Studies using knockout mice have been critical to understanding the mechanisms of the metabolic effects of SCFAs. GPR41
−/− mice were found to have a lower resting heart rate and UCP1 expression in brown fat, and their energy expenditure remained unchanged after propionate treatment [87]. Mice lacking GPR109A were obese and developed hepatic steatosis on a chow diet [101]. GPR43
mice were found to have a lower resting heart rate and UCP1 expression in brown fat, and their energy expenditure remained unchanged after propionate treatment [75]. Mice lacking GPR109A were obese and developed hepatic steatosis on a chow diet [89]. GPR43
−/− mice were found to be obese on a chow diet and had higher body weights than wild-type mice on an HFD [88]. This translated into impaired glucose tolerance and reduced sensitivity to exogenous insulin in clamp studies [88]. In contrast, overexpression of GPR43 in adipose tissue protected from HFD-induced obesity, lowered fasting blood glucose, increased energy expenditure and fat metabolism, and reduced liver triglycerides [88]. Treatment with antibiotics or housing animals in germ-free conditions completely blocked the effects of GPR43 deletion and overexpression, suggesting that these effects are microbiome-dependent [88].
mice were found to be obese on a chow diet and had higher body weights than wild-type mice on an HFD [76]. This translated into impaired glucose tolerance and reduced sensitivity to exogenous insulin in clamp studies [76]. In contrast, overexpression of GPR43 in adipose tissue protected from HFD-induced obesity, lowered fasting blood glucose, increased energy expenditure and fat metabolism, and reduced liver triglycerides [76]. Treatment with antibiotics or housing animals in germ-free conditions completely blocked the effects of GPR43 deletion and overexpression, suggesting that these effects are microbiome-dependent [76].
5. Molecular Mechanisms of Adverse Effects of Carbohydrate Intake
Diverse mechanisms have been proposed to mediate the molecular effects of carbohydrate intake. Rapid digestion of simple carbohydrates produces spikes of insulin secretion that cause a dip in blood glucose levels and stimulation of appetite [6]. Moreover, carbohydrate-induced insulin release may facilitate fat deposition by stimulating lipogenesis and inhibiting lipolysis [3]. The ketone or aldehyde moiety of carbohydrate molecules can react with the amino group of lysine in proteins or DNA bases, or with a free hydroxyl group of lipids to generate reactive oxygen species (ROS) [43]. Increased production of ROS has been linked to insulin resistance and pancreatic beta-cell dysfunction in diabetes [102]. Dihydroxyacetone phosphate and methylglyoxal produced from cellular glucose metabolism can also react with free amino groups found in proteins to form advanced glycation end products (AGEs) [6,103]. AGEs are widely reported to mediate complications of diabetes in several tissues [6].
Diverse mechanisms have been proposed to mediate the molecular effects of carbohydrate intake. Rapid digestion of simple carbohydrates produces spikes of insulin secretion that cause a dip in blood glucose levels and stimulation of appetite [4]. Moreover, carbohydrate-induced insulin release may facilitate fat deposition by stimulating lipogenesis and inhibiting lipolysis [1]. The ketone or aldehyde moiety of carbohydrate molecules can react with the amino group of lysine in proteins or DNA bases, or with a free hydroxyl group of lipids to generate reactive oxygen species (ROS) [31]. Increased production of ROS has been linked to insulin resistance and pancreatic beta-cell dysfunction in diabetes [90]. Dihydroxyacetone phosphate and methylglyoxal produced from cellular glucose metabolism can also react with free amino groups found in proteins to form advanced glycation end products (AGEs) [4][91]. AGEs are widely reported to mediate complications of diabetes in several tissues [4].
It has been suggested that the pro-lipogenic nature of fructose metabolism in the liver makes it more detrimental for metabolic health than other carbohydrates [6,43] (
It has been suggested that the pro-lipogenic nature of fructose metabolism in the liver makes it more detrimental for metabolic health than other carbohydrates [4][31] (
Figure 1). Around 50–75% of the fructose absorbed by the intestines is metabolised in the liver [104]. After entering the hepatocytes through GLUT2 and GLUT5 transporters, the enzyme ketohexokinase (KHK; also called fructokinase) phosphorylates fructose to fructose-1-phosphate [6,105]. The enzyme aldolase-B then converts fructose-1-phosphate into D-glyceraldehyde and dihydroxyacetone phosphate [6,105]. Further downstream metabolism of these three-carbon metabolites can lead to fatty acid synthesis via acetyl-CoA or generate a glycerol backbone of triglycerides [105]. Contrary to glucose, fructose metabolism in the liver is not tightly regulated by insulin signalling or by negative feedback from ATP and citrate, and this absence of feedback signals facilitates the potent induction of de novo lipogenesis (DNL) [6]. Comparison of DNL induction by high glucose vs. fructose intake for six days in humans showed a fractional DNL rate of 2% with glucose and up to 10% with fructose [43,106]. Studies in mice where a high-fat diet was supplemented with either a 30% fructose or glucose solution showed that both these monosaccharides activated the lipogenic factor ChREBP in the liver [107]. However, fructose additionally activated the lipogenic transcription factor SREBP1c and genes associated with fatty acid synthesis [107]. Glucose activates ChREBP, which leads to increased glycolysis and fatty acid synthesis [108]. Activation of the glucose-response activation conserved element (GRACE) domain of ChREBP by glucose metabolites leads to the binding of ChREBP to carbohydrate response element (ChoRE) sequences present on the promoter of DNL pathway genes such as
). Around 50–75% of the fructose absorbed by the intestines is metabolised in the liver [92]. After entering the hepatocytes through GLUT2 and GLUT5 transporters, the enzyme ketohexokinase (KHK; also called fructokinase) phosphorylates fructose to fructose-1-phosphate [4][93]. The enzyme aldolase-B then converts fructose-1-phosphate into D-glyceraldehyde and dihydroxyacetone phosphate [4][93]. Further downstream metabolism of these three-carbon metabolites can lead to fatty acid synthesis via acetyl-CoA or generate a glycerol backbone of triglycerides [93]. Contrary to glucose, fructose metabolism in the liver is not tightly regulated by insulin signalling or by negative feedback from ATP and citrate, and this absence of feedback signals facilitates the potent induction of de novo lipogenesis (DNL) [4]. Comparison of DNL induction by high glucose vs. fructose intake for six days in humans showed a fractional DNL rate of 2% with glucose and up to 10% with fructose [31][94]. Studies in mice where a high-fat diet was supplemented with either a 30% fructose or glucose solution showed that both these monosaccharides activated the lipogenic factor ChREBP in the liver [95]. However, fructose additionally activated the lipogenic transcription factor SREBP1c and genes associated with fatty acid synthesis [95]. Glucose activates ChREBP, which leads to increased glycolysis and fatty acid synthesis [96]. Activation of the glucose-response activation conserved element (GRACE) domain of ChREBP by glucose metabolites leads to the binding of ChREBP to carbohydrate response element (ChoRE) sequences present on the promoter of DNL pathway genes such as
Fas
,
Acc
and
Scd1 and increases their mRNA expression [109,110]. Fructose-induced hyperinsulinaemia and the resultant increase in insulin signalling increases the expression of DNL genes by activating SREBP-1c in the liver [111]. Mice with defects in the processing of SREBP-1c in ER and Golgi (required for SREBP-1c activation and its nuclear translocation) had markedly reduced insulin-induced DNL [112,113]. For example, deficiency of Scap, a protein that escorts SREBPs from ER to Golgi, reduced liver fat and triglyceridaemia in high-fat diet and high-sucrose diet models of rodent obesity [114]. Fructose-induced DNL results in the generation and secretion of very-low-density lipoprotein (VLDL) particles into the systemic circulation that contributes to hypertriglyceridaemia and adversely affects lipid profile [45]. In addition, compared with glucose solution, the supplementation of a high-fat diet with fructose solution reduced fatty acid oxidation in mice [115]. This was thought to be mediated by a fructose-induced increase in concentrations of hepatic malonyl-CoA (an inhibitor of fat oxidation), mitochondrial dysfunction characterised by reduced mitochondrial area, and increased inhibitory acetylation of fat oxidation pathway factors CPT1a and ACADL [115]. Overall, these changes in fat metabolism culminate in hepatic steatosis, increased visceral adiposity, and ectopic lipid deposition in liver and muscle tissue [42,44,45]. The toxic lipid species (ceramide and diacylglycerol) generated secondary to ectopic lipid accumulation are proposed to eventually inhibit insulin signalling, leading to insulin resistance [45].
and increases their mRNA expression [97][98]. Fructose-induced hyperinsulinaemia and the resultant increase in insulin signalling increases the expression of DNL genes by activating SREBP-1c in the liver [99]. Mice with defects in the processing of SREBP-1c in ER and Golgi (required for SREBP-1c activation and its nuclear translocation) had markedly reduced insulin-induced DNL [100][101]. For example, deficiency of Scap, a protein that escorts SREBPs from ER to Golgi, reduced liver fat and triglyceridaemia in high-fat diet and high-sucrose diet models of rodent obesity [102]. Fructose-induced DNL results in the generation and secretion of very-low-density lipoprotein (VLDL) particles into the systemic circulation that contributes to hypertriglyceridaemia and adversely affects lipid profile [33]. In addition, compared with glucose solution, the supplementation of a high-fat diet with fructose solution reduced fatty acid oxidation in mice [103]. This was thought to be mediated by a fructose-induced increase in concentrations of hepatic malonyl-CoA (an inhibitor of fat oxidation), mitochondrial dysfunction characterised by reduced mitochondrial area, and increased inhibitory acetylation of fat oxidation pathway factors CPT1a and ACADL [103]. Overall, these changes in fat metabolism culminate in hepatic steatosis, increased visceral adiposity, and ectopic lipid deposition in liver and muscle tissue [30][32][33]. The toxic lipid species (ceramide and diacylglycerol) generated secondary to ectopic lipid accumulation are proposed to eventually inhibit insulin signalling, leading to insulin resistance [33].
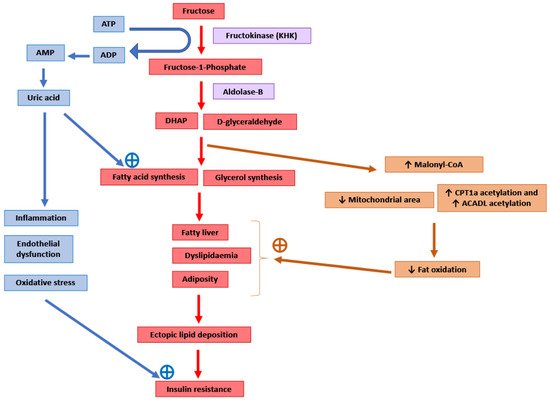
Potential molecular mechanisms mediating the adverse consequences of high fructose intake. DHAP: Dihydroxyacetone phosphate. CPT1a: Carnitine palmitoyltransferase 1 (hepatic isoform a). ACADL: Long-chain specific acyl-CoA dehydrogenase.
The enzyme KHK is the major mediator of metabolic outcomes of high fructose intake, and recent research has identified this enzyme as a potential therapeutic target for the management of obesity and fatty liver [104,115,116]. KHK exists in two isoforms: KHK-C and KHK-A. The highly active isoform “C” is expressed mainly in the liver, kidney and intestines, and metabolises the majority of the fructose absorbed from the diet [104,116]. The isoform “A” has widespread but low levels of tissue expression and a low affinity for fructose [104,116]. Mice provided with a 30% fructose solution to drink for 10 weeks as well as obese human subjects with non-alcoholic steatohepatitis (NASH) showed increased hepatic expression of KHK and downstream lipogenic genes
The enzyme KHK is the major mediator of metabolic outcomes of high fructose intake, and recent research has identified this enzyme as a potential therapeutic target for the management of obesity and fatty liver [92][103][104]. KHK exists in two isoforms: KHK-C and KHK-A. The highly active isoform “C” is expressed mainly in the liver, kidney and intestines, and metabolises the majority of the fructose absorbed from the diet [92][104]. The isoform “A” has widespread but low levels of tissue expression and a low affinity for fructose [92][104]. Mice provided with a 30% fructose solution to drink for 10 weeks as well as obese human subjects with non-alcoholic steatohepatitis (NASH) showed increased hepatic expression of KHK and downstream lipogenic genes
Acaca, Acly
and
Scd1 [107]. Knockdown of hepatic KHK by siRNA or combined deletion of KHK-A and KHK-C protected the mice from high fructose-induced obesity, fatty liver, glucose intolerance and insulin resistance [104,107,116,117]. The activity of KHK results in the breakdown of ATP to ADP and AMP, and this ATP depletion may result in an increase in phosphofructokinase (PFK) activity, leading to the increased utilisation of glucose for glycolysis and downstream DNL [118]. ATP degradation also activates AMP deaminase which converts AMP to inosine monophosphate, which is further converted into hypoxanthine, xanthine, and eventually into uric acid [6]. Uric acid is a pro-inflammatory metabolite that induces mitochondrial oxidative stress and inhibits aconitase in the citric acid cycle [6,119]. This stimulates DNL by the accumulation of citrate and the stimulation of ACLY and FASN enzymes [119]. Serum uric acid levels correlated positively in non-diabetic human subjects with the severity of hepatic steatosis [119]. Allopurinol, a xanthine oxidase inhibitor that blocks the conversion of xanthine to uric acid, reduced hepatic steatosis in a mouse model of metabolic syndrome [119]. Additionally, uric acid causes endothelial dysfunction that could lead to hypertension and insulin resistance [48].
[95]. Knockdown of hepatic KHK by siRNA or combined deletion of KHK-A and KHK-C protected the mice from high fructose-induced obesity, fatty liver, glucose intolerance and insulin resistance [92][95][104][105]. The activity of KHK results in the breakdown of ATP to ADP and AMP, and this ATP depletion may result in an increase in phosphofructokinase (PFK) activity, leading to the increased utilisation of glucose for glycolysis and downstream DNL [106]. ATP degradation also activates AMP deaminase which converts AMP to inosine monophosphate, which is further converted into hypoxanthine, xanthine, and eventually into uric acid [4]. Uric acid is a pro-inflammatory metabolite that induces mitochondrial oxidative stress and inhibits aconitase in the citric acid cycle [4][107]. This stimulates DNL by the accumulation of citrate and the stimulation of ACLY and FASN enzymes [107]. Serum uric acid levels correlated positively in non-diabetic human subjects with the severity of hepatic steatosis [107]. Allopurinol, a xanthine oxidase inhibitor that blocks the conversion of xanthine to uric acid, reduced hepatic steatosis in a mouse model of metabolic syndrome [107]. Additionally, uric acid causes endothelial dysfunction that could lead to hypertension and insulin resistance [36].
Sucrose (or fructose) are often added to high-fat rodent diets to model Western diets that are rich in fat and sugar [116]. This is because adding sucrose to a high-fat diet makes the metabolic impairment more severe [120,121]. In the liver, in addition to steatosis, this combination of sucrose and fat facilitates the induction of mild inflammation and fibrosis [116,122]. This indicates that fructose interacts with other nutrients in the diet to influence the metabolic phenotype. However, the mechanistic underpinnings of the interaction of fructose (and glucose) with fat and protein have not been examined in detail.
Sucrose (or fructose) are often added to high-fat rodent diets to model Western diets that are rich in fat and sugar [104]. This is because adding sucrose to a high-fat diet makes the metabolic impairment more severe [108][109]. In the liver, in addition to steatosis, this combination of sucrose and fat facilitates the induction of mild inflammation and fibrosis [104][110]. This indicates that fructose interacts with other nutrients in the diet to influence the metabolic phenotype. However, the mechanistic underpinnings of the interaction of fructose (and glucose) with fat and protein have not been examined in detail.
References
- Ludwig, D.S.; Willett, W.C.; Volek, J.S.; Neuhouser, M.L. Dietary fat: From foe to friend? Science 2018, 362, 764–770.
- Elia, M.; Cummings, J.H. Physiological aspects of energy metabolism and gastrointestinal effects of carbohydrates. Eur. J. Clin. Nutr. 2007, 61 (Suppl. 1), S40–S74.
- Westman, E.C. Is dietary carbohydrate essential for human nutrition? Am. J. Clin. Nutr. 2002, 75, 951–953.
- Kroemer, G.; Lopez-Otin, C.; Madeo, F.; de Cabo, R. Carbotoxicity-Noxious Effects of Carbohydrates. Cell 2018, 175, 605–614.
- Dehghan, M.; Mente, A.; Zhang, X.; Swaminathan, S.; Li, W.; Mohan, V.; Iqbal, R.; Kumar, R.; Wentzel-Viljoen, E.; Rosengren, A.; et al. Associations of fats and carbohydrate intake with cardiovascular disease and mortality in 18 countries from five continents (PURE): A prospective cohort study. Lancet 2017, 390, 2050–2062.
- Feinman, R.D.; Pogozelski, W.K.; Astrup, A.; Bernstein, R.K.; Fine, E.J.; Westman, E.C.; Accurso, A.; Frassetto, L.; Gower, B.A.; McFarlane, S.I.; et al. Dietary carbohydrate restriction as the first approach in diabetes management: Critical review and evidence base. Nutrition 2015, 31, 1–13.
- Mente, A.; Dehghan, M.; Rangarajan, S.; McQueen, M.; Dagenais, G.; Wielgosz, A.; Lear, S.; Li, W.; Chen, H.; Yi, S. Association of dietary nutrients with blood lipids and blood pressure in 18 countries: A cross-sectional analysis from the PURE study. Lancet Diabetes Endocrinol. 2017, 5, 774–787.
- Ludwig, D.S.; Hu, F.B.; Tappy, L.; Brand-Miller, J. Dietary carbohydrates: Role of quality and quantity in chronic disease. BMJ 2018, 361, k2340.
- Keenan, M.J.; Zhou, J.; Hegsted, M.; Pelkman, C.; Durham, H.A.; Coulon, D.B.; Martin, R.J. Role of resistant starch in improving gut health, adiposity, and insulin resistance. Adv. Nutr. 2015, 6, 198–205.
- Reynolds, A.; Mann, J.; Cummings, J.; Winter, N.; Mete, E.; Te Morenga, L. Carbohydrate quality and human health: A series of systematic reviews and meta-analyses. Lancet 2019, 393, 434–445.
- Choo, V.L.; Viguiliouk, E.; Blanco Mejia, S.; Cozma, A.I.; Khan, T.A.; Ha, V.; Wolever, T.M.S.; Leiter, L.A.; Vuksan, V.; Kendall, C.W.C.; et al. Food sources of fructose-containing sugars and glycaemic control: Systematic review and meta-analysis of controlled intervention studies. BMJ 2018, 363.
- Bray, G.A.; Nielsen, S.J.; Popkin, B.M. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am. J. Clin. Nutr. 2004, 79, 537–543.
- Blaut, M. Gut microbiota and energy balance: Role in obesity. Proc. Nutr. Soc. 2015, 74, 227–234.
- Consortium, I. Dietary Fibre and Incidence of Type 2 Diabetes in Eight European Countries: The EPIC-InterAct Study and a Meta-analysis of Prospective Studies; Springer: Berlin/Heidelberg, Germany, 2015.
- King, D.E.; Mainous III, A.G.; Lambourne, C.A. Trends in dietary fiber intake in the United States, 1999–2008. J. Acad. Nutr. Diet. 2012, 112, 642–648.
- Chen, G.X.; Zhou, J.W.; Liu, Y.L.; Lu, X.B.; Han, C.X.; Zhang, W.Y.; Xu, Y.H.; Yan, Y.M. Biosynthesis and Regulation of Wheat Amylose and Amylopectin from Proteomic and Phosphoproteomic Characterization of Granule-binding Proteins. Sci. Rep. 2016, 6, 33111.
- Aller, E.E.; Abete, I.; Astrup, A.; Martinez, J.A.; Baak, M.A.v. Starches, sugars and obesity. Nutrients 2011, 3, 341–369.
- Bhosale, S.H.; Rao, M.B.; Deshpande, V.V. Molecular and industrial aspects of glucose isomerase. Microbiol. Rev. 1996, 60, 280–300.
- Vos, M.B.; Kimmons, J.E.; Gillespie, C.; Welsh, J.; Blanck, H.M. Dietary fructose consumption among US children and adults: The Third National Health and Nutrition Examination Survey. Medscape J. Med. 2008, 10, 160.
- Stanhope, K.L.; Griffen, S.C.; Bair, B.R.; Swarbrick, M.M.; Keim, N.L.; Havel, P.J. Twenty-four-hour endocrine and metabolic profiles following consumption of high-fructose corn syrup-, sucrose-, fructose-, and glucose-sweetened beverages with meals. Am. J. Clin. Nutr. 2008, 87, 1194–1203.
- Lanaspa, M.A.; Ishimoto, T.; Li, N.; Cicerchi, C.; Orlicky, D.J.; Ruzycki, P.; Rivard, C.; Inaba, S.; Roncal-Jimenez, C.A.; Bales, E.S.; et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat. Commun. 2013, 4, 2434.
- Carden, T.J.; Carr, T.P. Food availability of glucose and fat, but not fructose, increased in the U.S. between 1970 and 2009: Analysis of the USDA food availability data system. Nutr. J. 2013, 12, 130.
- Elliott, S.S.; Keim, N.L.; Stern, J.S.; Teff, K.; Havel, P.J. Fructose, weight gain, and the insulin resistance syndrome. Am. J. Clin. Nutr. 2002, 76, 911–922.
- De Koning, L.; Malik, V.S.; Rimm, E.B.; Willett, W.C.; Hu, F.B. Sugar-sweetened and artificially sweetened beverage consumption and risk of type 2 diabetes in men. Am. J. Clin. Nutr. 2011, 93, 1321–1327.
- O’Connor, L.; Imamura, F.; Lentjes, M.A.; Khaw, K.-T.; Wareham, N.J.; Forouhi, N.G. Prospective associations and population impact of sweet beverage intake and type 2 diabetes, and effects of substitutions with alternative beverages. Diabetologia 2015, 58, 1474–1483.
- Imamura, F.; O’Connor, L.; Ye, Z.; Mursu, J.; Hayashino, Y.; Bhupathiraju, S.N.; Forouhi, N.G. Consumption of sugar sweetened beverages, artificially sweetened beverages, and fruit juice and incidence of type 2 diabetes: Systematic review, meta-analysis, and estimation of population attributable fraction. BMJ 2015, 351, h3576.
- Malik, V.S.; Popkin, B.M.; Bray, G.A.; Després, J.-P.; Willett, W.C.; Hu, F.B. Sugar sweetened beverages and risk of metabolic syndrome and type 2 diabetes: A meta-analysis. Diabetes Care 2010, 33, 2477–2483.
- Tappy, L.; Le, K.A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46.
- Stanhope, K.L. More pieces of the fructose puzzle. J. Intern. Med. 2017, 282, 202–204.
- Rippe, J.M.; Angelopoulos, T.J. Fructose-containing sugars and cardiovascular disease. Adv. Nutr. 2015, 6, 430–439.
- Lustig, R.H. Fructose: It’s “Alcohol Without the Buzz”; Oxford University Press: Oxford, UK, 2013.
- Lustig, R.H. Sickeningly Sweet: Does Sugar Cause Type 2 Diabetes? Yes. Can J. Diabetes 2016, 40, 282–286.
- Stanhope, K.L. Sugar consumption, metabolic disease and obesity: The state of the controversy. Crit. Rev. Clin. Lab. Sci. 2015.
- Te Morenga, L.; Mallard, S.; Mann, J. Dietary sugars and body weight: Systematic review and meta-analyses of randomised controlled trials and cohort studies. BMJ 2013, 346, e7492.
- Lean, M.E.; Te Morenga, L. Sugar and Type 2 diabetes. Br. Med. Bull. 2016, 120, 43–53.
- Rosset, R.; Surowska, A.; Tappy, L. Pathogenesis of Cardiovascular and Metabolic Diseases: Are Fructose-Containing Sugars More Involved Than Other Dietary Calories? Curr. Hypertens Rep. 2016, 18, 44.
- Olsen, N.J.; Heitmann, B.L. Intake of calorically sweetened beverages and obesity. Obes. Rev. 2009, 10, 68–75.
- Reid, M.; Hammersley, R.; Hill, A.J.; Skidmore, P. Long-term dietary compensation for added sugar: Effects of supplementary sucrose drinks over a 4-week period. Br. J. Nutr. 2007, 97, 193–203.
- Mattes, R.D. Dietary compensation by humans for supplemental energy provided as ethanol or carbohydrate in fluids. Physiol. Behav. 1996, 59, 179–187.
- Stice, E.; Burger, K.S.; Yokum, S. Relative ability of fat and sugar tastes to activate reward, gustatory, and somatosensory regions. Am. J. Clin. Nutr. 2013, 98, 1377–1384.
- Luo, S.; Monterosso, J.R.; Sarpelleh, K.; Page, K.A. Differential effects of fructose versus glucose on brain and appetitive responses to food cues and decisions for food rewards. Proc. Natl. Acad. Sci. USA 2015, 112, 6509–6514.
- Avena, N.M.; Rada, P.; Hoebel, B.G. Evidence for sugar addiction: Behavioral and neurochemical effects of intermittent, excessive sugar intake. Neurosci. Biobehav. Rev. 2008, 32, 20–39.
- Khan, T.A.; Sievenpiper, J.L. Controversies about sugars: Results from systematic reviews and meta-analyses on obesity, cardiometabolic disease and diabetes. Eur. J. Nutr. 2016, 55, 25–43.
- Rippe, J.M.; Kris Etherton, P.M. Fructose, sucrose, and high fructose corn syrup: Modern scientific findings and health implications. Adv. Nutr. 2012, 3, 739–740.
- Vega-López, S.; Venn, B.J.; Slavin, J.L. Relevance of the Glycemic Index and Glycemic Load for Body Weight, Diabetes, and Cardiovascular Disease. Nutrients 2018, 10, 1361.
- Radulian, G.; Rusu, E.; Dragomir, A.; Posea, M. Metabolic effects of low glycaemic index diets. Nutr. J. 2009, 8, 5.
- Brand-Miller, J.; Buyken, A.E. The Relationship between Glycemic Index and Health. Nutrients 2020, 12, 536.
- Geraghty, A.A.; Sexton-Oates, A.; O’Brien, E.C.; Alberdi, G.; Fransquet, P.; Saffery, R.; McAuliffe, F.M. A Low Glycaemic Index Diet in Pregnancy Induces DNA Methylation Variation in Blood of Newborns: Results from the ROLO Randomised Controlled Trial. Nutrients 2018, 10, 455.
- Yan, W.; Zhang, Y.; Wang, L.; Yang, W.; Li, C.; Wang, L.; Gu, P.; Xia, Y.; Yan, J.; Shen, Y.; et al. Maternal dietary glycaemic change during gestation influences insulin-related gene methylation in the placental tissue: A genome-wide methylation analysis. Genes Nutr. 2019, 14, 17.
- Pal, S.; Lim, S.; Egger, G. The effect of a low glycaemic index breakfast on blood glucose, insulin, lipid profiles, blood pressure, body weight, body composition and satiety in obese and overweight individuals: A pilot study. J. Am. Coll. Nutr. 2008, 27, 387–393.
- Krog-Mikkelsen, I.; Sloth, B.; Dimitrov, D.; Tetens, I.; Björck, I.; Flint, A.; Holst, J.J.; Astrup, A.; Elmståhl, H.; Raben, A. A low glycemic index diet does not affect postprandial energy metabolism but decreases postprandial insulinemia and increases fullness ratings in healthy women. J. Nutr. 2011, 141, 1679–1684.
- Aston, L.M.; Stokes, C.S.; Jebb, S.A. No effect of a diet with a reduced glycaemic index on satiety, energy intake and body weight in overweight and obese women. Int. J. Obes. 2008, 32, 160–165.
- Das, S.K.; Gilhooly, C.H.; Golden, J.K.; Pittas, A.G.; Fuss, P.J.; Cheatham, R.A.; Tyler, S.; Tsay, M.; McCrory, M.A.; Lichtenstein, A.H.; et al. Long-term effects of 2 energy-restricted diets differing in glycemic load on dietary adherence, body composition, and metabolism in CALERIE: A 1-y randomized controlled trial. Am. J. Clin. Nutr. 2007, 85, 1023–1030.
- Juanola-Falgarona, M.; Salas-Salvadó, J.; Ibarrola-Jurado, N.; Rabassa-Soler, A.; Díaz-López, A.; Guasch-Ferré, M.; Hernández-Alonso, P.; Balanza, R.; Bulló, M. Effect of the glycemic index of the diet on weight loss, modulation of satiety, inflammation, and other metabolic risk factors: A randomized controlled trial. Am. J. Clin. Nutr. 2014, 100, 27–35.
- Murakami, K.; Sasaki, S.; Okubo, H.; Takahashi, Y.; Hosoi, Y.; Itabashi, M. Dietary fiber intake, dietary glycemic index and load, and body mass index: A cross-sectional study of 3931 Japanese women aged 18-20 years. Eur. J. Clin. Nutr. 2007, 61, 986–995.
- Silva, F.M.; Steemburgo, T.; de Mello, V.D.; Tonding, S.F.; Gross, J.L.; Azevedo, M.J. High dietary glycemic index and low fiber content are associated with metabolic syndrome in patients with type 2 diabetes. J. Am. Coll Nutr. 2011, 30, 141–148.
- Abete, I.; Parra, D.; Martinez, J.A. Energy-restricted diets based on a distinct food selection affecting the glycemic index induce different weight loss and oxidative response. Clin. Nutr. 2008, 27, 545–551.
- Cheng, I.S.; Liao, S.F.; Liu, K.L.; Liu, H.Y.; Wu, C.L.; Huang, C.Y.; Mallikarjuna, K.; Smith, R.W.; Kuo, C.H. Effect of dietary glycemic index on substrate transporter gene expression in human skeletal muscle after exercise. Eur. J. Clin. Nutr. 2009, 63, 1404–1410.
- Mendez, M.A.; Covas, M.I.; Marrugat, J.; Vila, J.; Schröder, H. Glycemic load, glycemic index, and body mass index in Spanish adults. Am. J. Clin. Nutr. 2009, 89, 316–322.
- Milton, J.E.; Briche, B.; Brown, I.J.; Hickson, M.; Robertson, C.E.; Frost, G.S. Relationship of glycaemic index with cardiovascular risk factors: Analysis of the National Diet and Nutrition Survey for people aged 65 and older. Public Health Nutr. 2007, 10, 1321–1335.
- Castro-Quezada, I.; Artacho, R.; Molina-Montes, E.; Serrano, F.A.; Ruiz-López, M.D. Dietary glycaemic index and glycaemic load in a rural elderly population (60-74 years of age) and their relationship with cardiovascular risk factors. Eur. J. Nutr. 2015, 54, 523–534.
- Karl, J.P.; Roberts, S.B.; Schaefer, E.J.; Gleason, J.A.; Fuss, P.; Rasmussen, H.; Saltzman, E.; Das, S.K. Effects of carbohydrate quantity and glycemic index on resting metabolic rate and body composition during weight loss. Obesity 2015, 23, 2190–2198.
- Buscemi, S.; Cosentino, L.; Rosafio, G.; Morgana, M.; Mattina, A.; Sprini, D.; Verga, S.; Rini, G.B. Effects of hypocaloric diets with different glycemic indexes on endothelial function and glycemic variability in overweight and in obese adult patients at increased cardiovascular risk. Clin. Nutr. 2013, 32, 346–352.
- Philippou, E.; McGowan, B.M.; Brynes, A.E.; Dornhorst, A.; Leeds, A.R.; Frost, G.S. The effect of a 12-week low glycaemic index diet on heart disease risk factors and 24 h glycaemic response in healthy middle-aged volunteers at risk of heart disease: A pilot study. Eur. J. Clin. Nutr. 2008, 62, 145–149.
- Sichieri, R.; Moura, A.S.; Genelhu, V.; Hu, F.; Willett, W.C. An 18-mo randomized trial of a low-glycemic-index diet and weight change in Brazilian women. Am. J. Clin. Nutr. 2007, 86, 707–713.
- Villegas, R.; Liu, S.; Gao, Y.T.; Yang, G.; Li, H.; Zheng, W.; Shu, X.O. Prospective study of dietary carbohydrates, glycemic index, glycemic load, and incidence of type 2 diabetes mellitus in middle-aged Chinese women. Arch. Intern. Med. 2007, 167, 2310–2316.
- Sakurai, M.; Nakamura, K.; Miura, K.; Takamura, T.; Yoshita, K.; Morikawa, Y.; Ishizaki, M.; Kido, T.; Naruse, Y.; Suwazono, Y.; et al. Dietary glycemic index and risk of type 2 diabetes mellitus in middle-aged Japanese men. Metabolism 2012, 61, 47–55.
- Barclay, A.W.; Flood, V.M.; Rochtchina, E.; Mitchell, P.; Brand-Miller, J.C. Glycemic index, dietary fiber, and risk of type 2 diabetes in a cohort of older Australians. Diabetes Care 2007, 30, 2811–2813.
- Mosdøl, A.; Witte, D.R.; Frost, G.; Marmot, M.G.; Brunner, E.J. Dietary glycemic index and glycemic load are associated with high-density-lipoprotein cholesterol at baseline but not with increased risk of diabetes in the Whitehall II study. Am. J. Clin. Nutr. 2007, 86, 988–994.
- Jenkins, D.J.; Kendall, C.W.; McKeown-Eyssen, G.; Josse, R.G.; Silverberg, J.; Booth, G.L.; Vidgen, E.; Josse, A.R.; Nguyen, T.H.; Corrigan, S.; et al. Effect of a low-glycemic index or a high-cereal fiber diet on type 2 diabetes: A randomized trial. JAMA 2008, 300, 2742–2753.
- Wolever, T.M.; Gibbs, A.L.; Mehling, C.; Chiasson, J.L.; Connelly, P.W.; Josse, R.G.; Leiter, L.A.; Maheux, P.; Rabasa-Lhoret, R.; Rodger, N.W.; et al. The Canadian Trial of Carbohydrates in Diabetes (CCD), a 1-y controlled trial of low-glycemic-index dietary carbohydrate in type 2 diabetes: No effect on glycated hemoglobin but reduction in C-reactive protein. Am. J. Clin. Nutr. 2008, 87, 114–125.
- Bantle, J.P. Is fructose the optimal low glycemic index sweetener? Nestle Nutr. Workshop Ser. Clin. Perform Programme 2006, 11, 83–95.
- Livesey, G.; Taylor, R.; Livesey, H.F.; Buyken, A.E.; Jenkins, D.J.A.; Augustin, L.S.A.; Sievenpiper, J.L.; Barclay, A.W.; Liu, S.; Wolever, T.M.S.; et al. Dietary Glycemic Index and Load and the Risk of Type 2 Diabetes: A Systematic Review and Updated Meta-Analyses of Prospective Cohort Studies. Nutrients 2019, 11, 1280.
- Jenkins, D.J.A.; Dehghan, M.; Mente, A.; Bangdiwala, S.I.; Rangarajan, S.; Srichaikul, K.; Mohan, V.; Avezum, A.; Díaz, R.; Rosengren, A.; et al. Glycemic Index, Glycemic Load, and Cardiovascular Disease and Mortality. N. Engl. J. Med. 2021, 384, 1312–1322.
- Kimura, I.; Inoue, D.; Maeda, T.; Hara, T.; Ichimura, A.; Miyauchi, S.; Kobayashi, M.; Hirasawa, A.; Tsujimoto, G. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc. Natl. Acad. Sci. USA 2011, 108, 8030–8035.
- Kimura, I.; Ozawa, K.; Inoue, D.; Imamura, T.; Kimura, K.; Maeda, T.; Terasawa, K.; Kashihara, D.; Hirano, K.; Tani, T.; et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun. 2013, 4, 1829.
- Bindels, L.B.; Walter, J.; Ramer-Tait, A.E. Resistant starches for the management of metabolic diseases. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 559–565.
- Kieffer, D.A.; Piccolo, B.D.; Marco, M.L.; Kim, E.B.; Goodson, M.L.; Keenan, M.J.; Dunn, T.N.; Knudsen, K.E.; Martin, R.J.; Adams, S.H. Mice Fed a High-Fat Diet Supplemented with Resistant Starch Display Marked Shifts in the Liver Metabolome Concurrent with Altered Gut Bacteria. J. Nutr. 2016, 146, 2476–2490.
- Koay, Y.C.; Wali, J.A.; Luk, A.W.S.; Macia, L.; Cogger, V.C.; Pulpitel, T.J.; Wahl, D.; Solon-Biet, S.M.; Holmes, A.; Simpson, S.J.; et al. Ingestion of resistant starch by mice markedly increases microbiome-derived metabolites. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 8033–8042.
- Abildgaard, A.; Elfving, B.; Hokland, M.; Wegener, G.; Lund, S. The microbial metabolite indole-3-propionic acid improves glucose metabolism in rats, but does not affect behaviour. Arch. Physiol. Biochem. 2018, 124, 306–312.
- Robertson, M.D.; Bickerton, A.S.; Dennis, A.L.; Vidal, H.; Frayn, K.N. Insulin-sensitizing effects of dietary resistant starch and effects on skeletal muscle and adipose tissue metabolism. Am. J. Clin. Nutr. 2005, 82, 559–567.
- Canfora, E.E.; Jocken, J.W.; Blaak, E.E. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 2015, 11, 577–591.
- den Besten, G.; Bleeker, A.; Gerding, A.; van Eunen, K.; Havinga, R.; van Dijk, T.H.; Oosterveer, M.H.; Jonker, J.W.; Groen, A.K.; Reijngoud, D.J.; et al. Short-Chain Fatty Acids Protect Against High-Fat Diet-Induced Obesity via a PPARgamma-Dependent Switch From Lipogenesis to Fat Oxidation. Diabetes 2015, 64, 2398–2408.
- Lin, H.V.; Frassetto, A.; Kowalik, E.J., Jr.; Nawrocki, A.R.; Lu, M.M.; Kosinski, J.R.; Hubert, J.A.; Szeto, D.; Yao, X.; Forrest, G.; et al. Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS ONE 2012, 7, e35240.
- Lu, Y.; Fan, C.; Li, P.; Lu, Y.; Chang, X.; Qi, K. Short Chain Fatty Acids Prevent High-fat-diet-induced Obesity in Mice by Regulating G Protein-coupled Receptors and Gut Microbiota. Sci. Rep. 2016, 6, 37589.
- Li, H.; Gao, Z.; Zhang, J.; Ye, X.; Xu, A.; Ye, J.; Jia, W. Sodium butyrate stimulates expression of fibroblast growth factor 21 in liver by inhibition of histone deacetylase 3. Diabetes 2012, 61, 797–806.
- Kondo, T.; Kishi, M.; Fushimi, T.; Ugajin, S.; Kaga, T. Vinegar intake reduces body weight, body fat mass, and serum triglyceride levels in obese Japanese subjects. Biosci. Biotechnol. Biochem. 2009, 73, 1837–1843.
- Wanders, D.; Graff, E.C.; Judd, R.L. Effects of high fat diet on GPR109A and GPR81 gene expression. Biochem. Biophys. Res. Commun. 2012, 425, 278–283.
- Jadeja, R.N.; Jones, M.A.; Fromal, O.; Powell, F.L.; Khurana, S.; Singh, N.; Martin, P.M. Loss of GPR109A/HCAR2 induces aging-associated hepatic steatosis. Aging 2019, 11, 386–400.
- Kaneto, H.; Katakami, N.; Matsuhisa, M.; Matsuoka, T.A. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediat. Inflamm. 2010, 2010, 453892.
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Methylglyoxal, the dark side of glycolysis. Front. Neurosci. 2015, 9, 23.
- Marek, G.; Pannu, V.; Shanmugham, P.; Pancione, B.; Mascia, D.; Crosson, S.; Ishimoto, T.; Sautin, Y.Y. Adiponectin resistance and proinflammatory changes in the visceral adipose tissue induced by fructose consumption via ketohexokinase-dependent pathway. Diabetes 2015, 64, 508–518.
- Jurgens, H.; Haass, W.; Castaneda, T.R.; Schurmann, A.; Koebnick, C.; Dombrowski, F.; Otto, B.; Nawrocki, A.R.; Scherer, P.E.; Spranger, J.; et al. Consuming fructose-sweetened beverages increases body adiposity in mice. Obes. Res. 2005, 13, 1146–1156.
- Faeh, D.; Minehira, K.; Schwarz, J.-M.; Periasamy, R.; Park, S.; Tappy, L. Effect of fructose overfeeding and fish oil administration on hepatic de novo lipogenesis and insulin sensitivity in healthy men. Diabetes 2005, 54, 1907–1913.
- Softic, S.; Gupta, M.K.; Wang, G.X.; Fujisaka, S.; O’Neill, B.T.; Rao, T.N.; Willoughby, J.; Harbison, C.; Fitzgerald, K.; Ilkayeva, O.; et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J. Clin. Investig. 2017, 127, 4059–4074.
- Linden, A.G.; Li, S.; Choi, H.Y.; Fang, F.; Fukasawa, M.; Uyeda, K.; Hammer, R.E.; Horton, J.D.; Engelking, L.J.; Liang, G. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice. J. Lipid Res. 2018, 59, 475–487.
- Kawaguchi, T.; Takenoshita, M.; Kabashima, T.; Uyeda, K. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc. Natl. Acad. Sci. USA 2001, 98, 13710–13715.
- Ortega-Prieto, P.; Postic, C. Carbohydrate Sensing Through the Transcription Factor ChREBP. Front. Genet. 2019, 10, 472.
- Shao, W.; Espenshade, P.J. Expanding roles for SREBP in metabolism. Cell Metab. 2012, 16, 414–419.
- Matsuda, M.; Korn, B.S.; Hammer, R.E.; Moon, Y.A.; Komuro, R.; Horton, J.D.; Goldstein, J.L.; Brown, M.S.; Shimomura, I. SREBP cleavage-activating protein (SCAP) is required for increased lipid synthesis in liver induced by cholesterol deprivation and insulin elevation. Genes Dev. 2001, 15, 1206–1216.
- Yang, J.; Goldstein, J.L.; Hammer, R.E.; Moon, Y.A.; Brown, M.S.; Horton, J.D. Decreased lipid synthesis in livers of mice with disrupted Site-1 protease gene. Proc. Natl. Acad. Sci. USA 2001, 98, 13607–13612.
- Moon, Y.A.; Liang, G.; Xie, X.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Brown, M.S.; Goldstein, J.L.; Horton, J.D. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab. 2012, 15, 240–246.
- Softic, S.; Meyer, J.G.; Wang, G.X.; Gupta, M.K.; Batista, T.M.; Lauritzen, H.; Fujisaka, S.; Serra, D.; Herrero, L.; Willoughby, J.; et al. Dietary Sugars Alter Hepatic Fatty Acid Oxidation via Transcriptional and Post-translational Modifications of Mitochondrial Proteins. Cell Metab. 2019, 30, 735–753.e734.
- Ishimoto, T.; Lanaspa, M.A.; Rivard, C.J.; Roncal-Jimenez, C.A.; Orlicky, D.J.; Cicerchi, C.; McMahan, R.H.; Abdelmalek, M.F.; Rosen, H.R.; Jackman, M.R.; et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology 2013, 58, 1632–1643.
- Ishimoto, T.; Lanaspa, M.A.; Le, M.T.; Garcia, G.E.; Diggle, C.P.; Maclean, P.S.; Jackman, M.R.; Asipu, A.; Roncal-Jimenez, C.A.; Kosugi, T.; et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 4320–4325.
- Goncalves, M.D.; Lu, C.; Tutnauer, J.; Hartman, T.E.; Hwang, S.-K.; Murphy, C.J.; Pauli, C.; Morris, R.; Taylor, S.; Bosch, K. High-fructose corn syrup enhances intestinal tumor growth in mice. Science 2019, 363, 1345–1349.
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.-J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: Potential role in fructose-dependent and-independent fatty liver. J. Biol. Chem. 2012, 287, 40732–40744.
- Bortolin, R.C.; Vargas, A.R.; Gasparotto, J.; Chaves, P.R.; Schnorr, C.E.; Martinello, K.B.; Silveira, A.K.; Rabelo, T.K.; Gelain, D.P.; Moreira, J.C.F. A new animal diet based on human Western diet is a robust diet-induced obesity model: Comparison to high-fat and cafeteria diets in term of metabolic and gut microbiota disruption. Int. J. Obes. 2018, 42, 525–534.
- Surwit, R.; Feinglos, M.; Rodin, J.; Sutherland, A.; Petro, A.; Opara, E.; Kuhn, C.; Rebuffe-Scrive, M. Differential effects of fat and sucrose on the development of obesity and diabetes in C57BL/6J and AJ mice. Metabolism 1995, 44, 645–651.
- Farrell, G.; Schattenberg, J.M.; Leclercq, I.; Yeh, M.M.; Goldin, R.; Teoh, N.; Schuppan, D. Mouse Models of Nonalcoholic Steatohepatitis: Toward Optimization of Their Relevance to Human Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2241–2257.