B cells are key players in this relationship because activated and differentiated B cells produce secretory immunoglobulin A (sIgA), which binds commensal bacteria to preserve a healthy microbial ecosystem.
B cells are key players in this relationship because activated and differentiated B cells produce secretory immunoglobulin A (sIgA), which binds commensals to preserve a healthy microbial ecosystem.
- microbiota
- dysbiosis
- autoimmunity
- GALT
- Peyer’s patches
1. Introduction
B cells play a pivotal role in the dialogue between mucosal microbiota and the host´s immune system due to their capacity to differentiate into antibody-secreting cells that produce a substantial amount of IgA antibodies that are released within the mucosa. IgA dimers and J-chain antibody complexes produced in the lamina propria are transported through the epithelial cells after binding the poly-Ig-receptor (pIgR) on the basolateral side of the epithelial cells [1]. The IgA B-cell response is fundamental to maintain a healthy microbiota ecosystem; therefore, any modification in the IgA response will impact the microbial abundance and diversity in the gut mucosa leading to dysbiosis. This dysbiosis may lead to autoimmune inflammation under the proper conditions [2]. At the same time, gut commensals determine the type of IgA response: pathobionts of commensals with invasive properties induce T cell-dependent IgA responses, while the vast majority of bacteria induce T cell-independent IgA responses [3]. The origin of autoimmunity has long been suggested to be the cross-recognition of antigens on microorganisms, whether bacteria or viruses, and host antigens by molecular mimicry. Certainly, some mimotopes have been found in commensal bacteria that have been suggested to participate in the induction of autoimmune pathogenesis [4][5], as well as the production of antibodies to host antigens [6]. Apart from the local humoral response in the gut mucosa, the passage of bacterial components and whole bacteria, even in the steady state, provokes a systemic antibody response beyond the gut mucosa that results in circulating antibodies to gut resident bacteria, which increases in case of inflammation as the intestinal barrier becomes more permeable [7]. Besides producing antibodies, B cells can also serve as antigen-presenting cells (APC) and can produce substantial amounts of cytokines. IL-10 producing B cells, also known as regulatory B cells (Bregs), have the capacity to suppress autoimmune inflammation. The induction and differentiation of Bregs depend on signaling through innate immune receptors. Thus, it is not surprising that gut resident bacteria or bacterial metabolites are able to induce abundant production of IL-10 in B cells with anti-inflammatory activity in autoimmunity [8]. Moreover, bacterial metabolites can also suppress the production of inflammatory cytokines by B cells [9].
B cells play a pivotal role in the dialogue between mucosal microbiota and the host´s immune system due to their capacity to differentiate into antibody-secreting cells that produce a substantial amount of IgA antibodies that are released within the mucosa. IgA dimers and J-chain antibody complexes produced in the lamina propria are transported through the epithelial cells after binding the poly-Ig-receptor (pIgR) on the basolateral side of the epithelial cells [1]. The IgA B-cell response is fundamental to maintain a healthy microbiota ecosystem; therefore, any modification in the IgA response will impact the microbial abundance and diversity in the gut mucosa leading to dysbiosis. This dysbiosis may lead to autoimmune inflammation under the proper conditions [2]. At the same time, gut commensals determine the type of IgA response: pathobionts of commensals with invasive properties induce T cell-dependent IgA responses, while the vast majority of bacteria induce T cell-independent IgA responses [3]. The origin of autoimmunity has long been suggested to be the cross-recognition of antigens on microorganisms, whether bacteria or viruses, and host antigens by molecular mimicry. Certainly, some mimotopes have been found in commensal bacteria that have been suggested to participate in the induction of autoimmune pathogenesis [4,5], as well as the production of antibodies to host antigens [6]. Apart from the local humoral response in the gut mucosa, the passage of bacterial components and whole bacteria, even in the steady state, provokes a systemic antibody response beyond the gut mucosa that results in circulating antibodies to gut resident bacteria, which increases in case of inflammation as the intestinal barrier becomes more permeable [7]. Besides producing antibodies, B cells can also serve as antigen-presenting cells (APC) and can produce substantial amounts of cytokines. IL-10 producing B cells, also known as regulatory B cells (Bregs), have the capacity to suppress autoimmune inflammation. The induction and differentiation of Bregs depend on signaling through innate immune receptors. Thus, it is not surprising that gut resident bacteria or bacterial metabolites are able to induce abundant production of IL-10 in B cells with anti-inflammatory activity in autoimmunity [8]. Moreover, bacterial metabolites can also suppress the production of inflammatory cytokines by B cells [9].
2. B-Cell Lymphopoiesis and B-Cell Differentiation
The rising of B cells, a branch of lymphopoiesis, takes place in the bone marrow, a specialized primary lymphoid tissue. The cellular composition and specialized microenvironment of this organ allow the common lymphoid progenitor cell to give rise to B cells, ready to encounter their antigen. This possibility is given by a net of specialized non-lymphoid stromal cells and the surrounding osteoblasts located in intimate contact with the B-cell precursors and the to be, B cells [10][11][12]. As a consequence of this organization, the contribution supplied by stromal cells and osteoblasts is two-fold. First, establish adhesion contacts with the developing lymphocytes through cell-adhesion molecules and their ligands. Second, secrete cytokines and chemokines that can control differentiation and proliferation processes [13][14].
The rising of B cells, a branch of lymphopoiesis, takes place in the bone marrow, a specialized primary lymphoid tissue. The cellular composition and specialized microenvironment of this organ allow the common lymphoid progenitor cell to give rise to B cells, ready to encounter their antigen. This possibility is given by a net of specialized non-lymphoid stromal cells and the surrounding osteoblasts located in intimate contact with the B-cell precursors and the to be, B cells [10,11,12]. As a consequence of this organization, the contribution supplied by stromal cells and osteoblasts is two-fold. First, establish adhesion contacts with the developing lymphocytes through cell-adhesion molecules and their ligands. Second, secrete cytokines and chemokines that can control differentiation and proliferation processes [13,14].
In this context, to achieve the stage of the common lymphoid progenitor from stem cells, it is mandatory that the expression of the cell-surface receptor tyrosine kinase FLT3 and the transcription factor PU.1, together, induce the expression of the IL-7 receptor [15][16][17]. For the next step, osteoblasts and, to a lesser extent, stromal cells, produce IL-7, a cytokine essential for the growth and survival of developing B cells in mice. This necessary role has been exposed in experiments using mice that lack either IL-7, its receptor (IL-7R), or any of the molecules implicated in the IL-7R signaling cascade, where all tested animals exhibited a severe block in B-cell development [14][18].
In this context, to achieve the stage of the common lymphoid progenitor from stem cells, it is mandatory that the expression of the cell-surface receptor tyrosine kinase FLT3 and the transcription factor PU.1, together, induce the expression of the IL-7 receptor [15,16,17]. For the next step, osteoblasts and, to a lesser extent, stromal cells, produce IL-7, a cytokine essential for the growth and survival of developing B cells in mice. This necessary role has been exposed in experiments using mice that lack either IL-7, its receptor (IL-7R), or any of the molecules implicated in the IL-7R signaling cascade, where all tested animals exhibited a severe block in B-cell development [14,18].
The commitment of the B lineage is achieved along this process and is then when the early pro-B stage is defined. If this state is successfully accomplished, the genes Rag1 and Rag2 are expressed, initiating V(D)J recombination assembly [19][20][21][22], starting Ig heavy-chain rearrangement. This, together with the expression of the transcription factor Pax5, the surface molecule CD19, and the rearrangement of the Ig light chain, gives place to the late appearance of the pro-B cells [23][24]. When the µ heavy-chain locus is fully synthesized, the pre-B-cell receptor (pre-BCR) is complete, and the cell overtakes a tolerance checkpoint rendering it not to recognize self-structures, entering the pre-B state. This change carries an attenuation in IL-7R signaling in order to allow the cell to evolve into further stages [25][26]. These cells will be able to emerge from the bone marrow into the periphery once the pre-BCR couples with the rearranged light chain, allowing the IgM to be expressed on the cell surface (and, if needed, to be secreted as soluble IgM, sIgM) and becoming hence an immature B cell characterized by the expression of high levels of sIgM and low IgD [27][28]. When B cells enter the periphery, they undergo a final maturation step, when they encounter peripheral self-antigens and undergo tolerance creating a repertoire of B cells whose BCR activates following the recognition of foreign antigens.
The commitment of the B lineage is achieved along this process and is then when the early pro-B stage is defined. If this state is successfully accomplished, the genes Rag1 and Rag2 are expressed, initiating V(D)J recombination assembly [19,20,21,22], starting Ig heavy-chain rearrangement. This, together with the expression of the transcription factor Pax5, the surface molecule CD19, and the rearrangement of the Ig light chain, gives place to the late appearance of the pro-B cells [23,24]. When the µ heavy-chain locus is fully synthesized, the pre-B-cell receptor (pre-BCR) is complete, and the cell overtakes a tolerance checkpoint rendering it not to recognize self-structures, entering the pre-B state. This change carries an attenuation in IL-7R signaling in order to allow the cell to evolve into further stages [25,26]. These cells will be able to emerge from the bone marrow into the periphery once the pre-BCR couples with the rearranged light chain, allowing the IgM to be expressed on the cell surface (and, if needed, to be secreted as soluble IgM, sIgM) and becoming hence an immature B cell characterized by the expression of high levels of sIgM and low IgD [27,28]. When B cells enter the periphery, they undergo a final maturation step, when they encounter peripheral self-antigens and undergo tolerance creating a repertoire of B cells whose BCR activates following the recognition of foreign antigens.
It is known that B-cell development taking place during and after late fetal life is different from that occurring early in fetal ontogeny [29][30]. The process maintains the same general structure and stages of B-cell differentiation, but cells that develop from these early fetal progenitors give rise to lymphocytes populating mucosal and epithelial tissues involved in innate immune responses. In the adult lymphoid tissues, this population, known as B-1 B cells, represents a minor percentage in secondary lymphoid organs [29][31][32]. Up to this point, differentiation is antigen-independent [33][34].
It is known that B-cell development taking place during and after late fetal life is different from that occurring early in fetal ontogeny [29,30]. The process maintains the same general structure and stages of B-cell differentiation, but cells that develop from these early fetal progenitors give rise to lymphocytes populating mucosal and epithelial tissues involved in innate immune responses. In the adult lymphoid tissues, this population, known as B-1 B cells, represents a minor percentage in secondary lymphoid organs [29,31,32]. Up to this point, differentiation is antigen-independent [33,34].
The small intestine lamina propria (SILP) is, besides the bone marrow, a site for BCR diversification and B-cell development. Particularly during a small window at mice’s early life, RAG2-expressing pro- and pre-B cells undergo VDJ recombination in the SILP, and immature B cells undergo BCR editing [35]. RAG2+ B cells increase after colonization by gut commensals. Developing B cells expressing the TDT enzyme, important in the addition of nucleotides in the VH genes, localize mainly opposite to the luminal surface. In addition, the VH repertoire was found to be similar in BM and SILP developing B cells, but the V repertoire differed, possibly influenced by components of the commensal microbes, because microbial colonization exclusively increased the expression of immunoglobulin kappa light chain in SILP B cells.
To reach this stage, B cells need to activate, entering the antigen-dependent recognition phase [36]. To achieve this, B cells follow two paths: 1) the encounter of a T-independent antigen in the extrafollicular areas of the lymph nodes and spleen, a process that induces maturation of B cells and their transformation into IgM-secreting plasma cells. This subset of B cells is mainly characterized by low expression of the activation-induced cytidine deaminase (AICDA or AID), giving place to plasma cells with little class switch recombination [37][38] (
To reach this stage, B cells need to activate, entering the antigen-dependent recognition phase [36]. To achieve this, B cells follow two paths: 1) the encounter of a T-independent antigen in the extrafollicular areas of the lymph nodes and spleen, a process that induces maturation of B cells and their transformation into IgM-secreting plasma cells. This subset of B cells is mainly characterized by low expression of the activation-induced cytidine deaminase (AICDA or AID), giving place to plasma cells with little class switch recombination [37,38] (
).
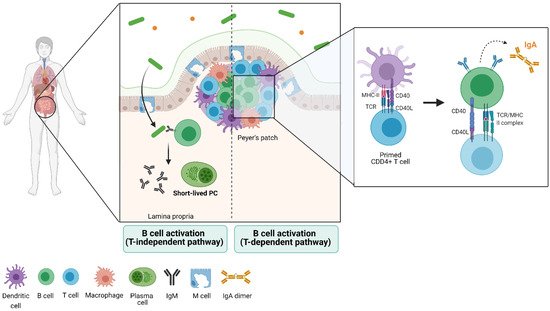
Figure 1.
In the second path, naïve B cells can enter the follicular areas and closely interact with antigen-presenting T cells, where they activate in a T-dependent manner, proliferating and creating germinal centers (GCs) [36][39][40]. Within these structures, B cells express BCL6 (transcription factor exclusive of this stage) and AID, which lead to the formation of plasma cells that undergo somatic hypermutation (SH) and class switch recombination (CSR) [36][41][42]. The clonal selection that takes place in GCs has been widely studied, as they usually form transiently in lymphoid organs in response to infection and immunization (
In the second path, naïve B cells can enter the follicular areas and closely interact with antigen-presenting T cells, where they activate in a T-dependent manner, proliferating and creating germinal centers (GCs) [36,39,40]. Within these structures, B cells express BCL6 (transcription factor exclusive of this stage) and AID, which lead to the formation of plasma cells that undergo somatic hypermutation (SH) and class switch recombination (CSR) [36,41,42]. The clonal selection that takes place in GCs has been widely studied, as they usually form transiently in lymphoid organs in response to infection and immunization (
).
In lymphoid organs associated with the gut, however, germinal centers are invariably present [43][44]. In this context, the differentiation into memory B cells is primarily generated through T-independent responses [45]. Overall, IgM-producing plasma cells (PCs) may tend to live a few days in mice and humans (short-lived PC) or up to a year in mice and decades in humans (long-lived PC) [46][47][48]. The process to a PC state is mainly determined by the repression of the transcription factors that establish B-cell identity during its development, Pax5 and BCL6 [23][24][49]. The expression of Blimp-1, a transcriptional repressor, is required to achieve full PC differentiation. In mice deficient for the gene encoding Blimp-1,
In lymphoid organs associated with the gut, however, germinal centers are invariably present [43,44]. In this context, the differentiation into memory B cells is primarily generated through T-independent responses [45]. Overall, IgM-producing plasma cells (PCs) may tend to live a few days in mice and humans (short-lived PC) or up to a year in mice and decades in humans (long-lived PC) [46,47,48]. The process to a PC state is mainly determined by the repression of the transcription factors that establish B-cell identity during its development, Pax5 and BCL6 [23,24,49]. The expression of Blimp-1, a transcriptional repressor, is required to achieve full PC differentiation. In mice deficient for the gene encoding Blimp-1,
Prdm1−/−, early pre-plasmablasts are still present [23][50].
, early pre-plasmablasts are still present [23,50].
Only a small percentage of immature B cells leaving the bone marrow and entering systemic circulation become fully mature B cells. This selection process is driven by the limited number of lymphoid follicles in which mature B cells can locate, become activated, and proliferate [47][51][52][53]. These survival niches for PCs are located in the bone marrow, gut-associated lymphoid tissues (GALT), lymph nodes, and spleen [53][54][55][56].
Only a small percentage of immature B cells leaving the bone marrow and entering systemic circulation become fully mature B cells. This selection process is driven by the limited number of lymphoid follicles in which mature B cells can locate, become activated, and proliferate [47,51,52,53]. These survival niches for PCs are located in the bone marrow, gut-associated lymphoid tissues (GALT), lymph nodes, and spleen [53,54,55,56].