Cutaneous Melanoma (CM), arising from pigment-producing melanocytes in the skin, is an aggressive cancer with high metastatic potential. While cutaneous melanoma represents only a fraction of all skin cancers (<5%), it accounts for most skin-cancer-related deaths worldwide. Immune checkpoint inhibition has been the first therapeutic approach to significantly benefit patient survival after treatment. Nevertheless, the immunosuppressive tumor microenvironment and the intrinsic and acquired treatment resistance of melanoma remain crucial challenges. Combining local and systemic treatment offers the potential to augment therapeutic response and overcome resistance, although, complex drug combinations can harbor an increased risk of immune-related adverse events.
- skin cancer
- melanoma
- cancer therapy
- immunotherapy
- targeted therapy
- intratumoral therapy
- combination therapy
1. Introduction
Cancer of the skin is considered a growing epidemic among Caucasians in the western world, with an alarming increase of non-melanoma skin cancer and cutaneous melanoma (CM) incidence rates of up to 44% over the last decade [1][2][3]. Moreover, increased accumulated exposure to ultraviolet (UV) radiation of the sun over lifespan due to growing life expectancy and the global climate crisis are considered major catalysts of increased incidence and mortality of skin cancer. Whilst CM is still most common in the male population aged 50–70 years, rates of CM in young adults, specifically in young women, are constantly increasing over the last years. Although recently approved novel targeted and immunotherapeutic approaches for CM treatment have been able to contain mortality rates of melanoma patients, it is predicted that nearly half a million people will be diagnosed with CM by 2040 with an increase in incidence of 62% and an increase of mortality of up to 74% [4][5][6]. In contrast to non-melanoma skin cancer (0.69) the age-standardized annual mortality rate for CM is about 1.5 (per 100,000 population) in the United States. These alarming numbers make the development of novel targeted therapeutic options and constant adjustments of the current state-of-the-art regimens essential in successful CM treatment. Several combinations involving immune checkpoint inhibition (ICI), targeted therapy through mutant-BRAF inhibition, intratumoral application of immunomodulators, oncolytic viruses, and anti-angiogenic approaches are being trialed to prime the anti-tumor response, enhance the sensitivity of CM cells to therapeutic interventions, and overcome therapy resistance of mutant cancer cells (Figure 1).
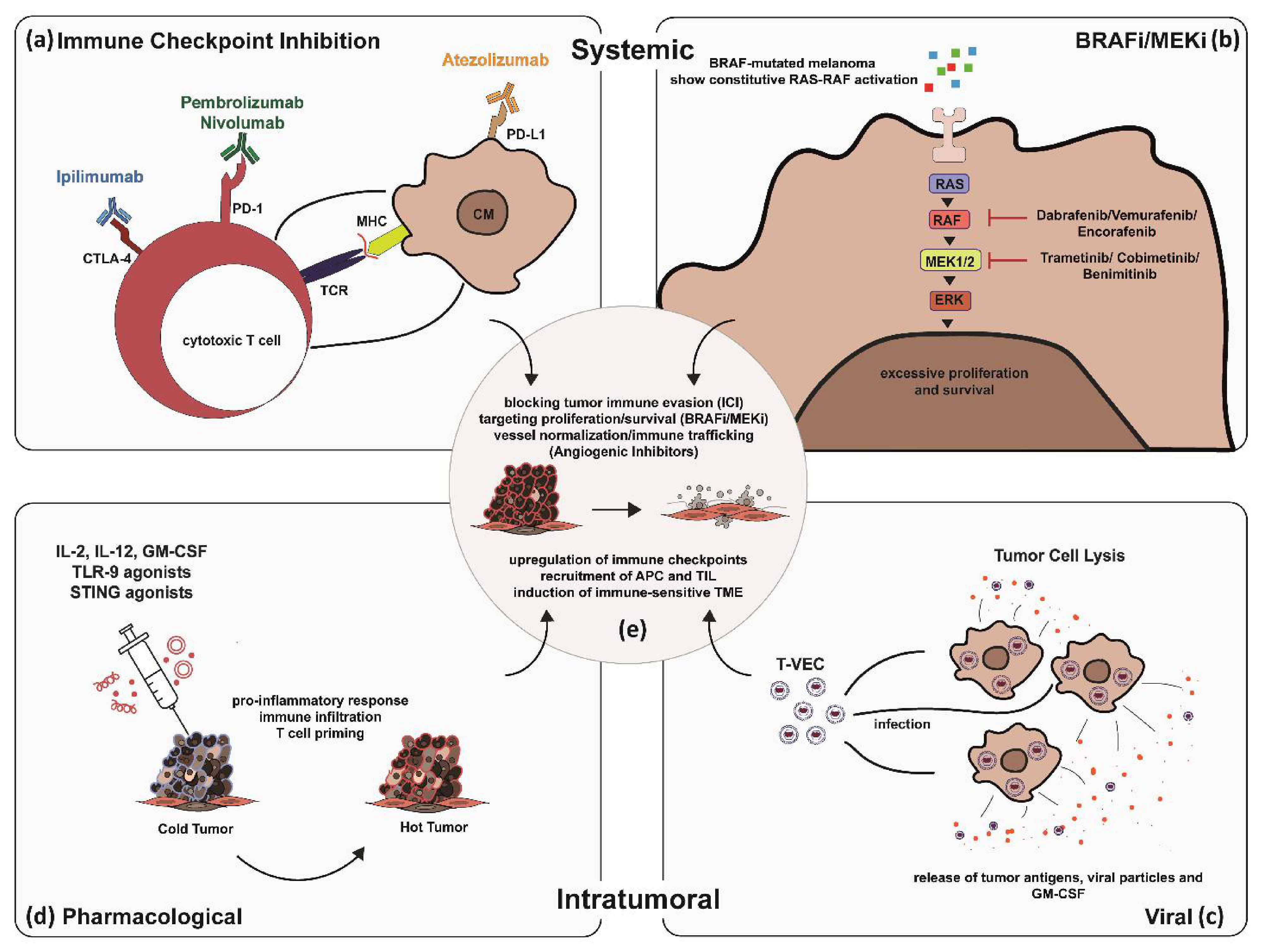
Figure 1. Synergistic effects of combined systemic and intratumoral therapy in skin cancer treatment exemplified by CM. Systemic immune checkpoint inhibition (a–e): Blocking of immune checkpoints on cytotoxic T Lymphocytes (CTL) and CM cells disrupting tumor immune evasion (a). Systemic BRAFi/MEKi: Blocking overactivation of RAS-RAF signaling pathway in BRAF-mutated melanoma by BRAF and MEK inhibitors (b). Intratumoral application of genetically engineered oncolytic viral particles to express checkpoint inhibitors on CM cells, force production and release of tumor antigens and GM-CSF to prime T cell immune response and induce tumor cell lysis (c). Intratumoral pharmacological application of IL-2, IL12, GM-CSF, TLR-9 and STING agonists: Priming locally advanced CM to become immunogenic (d).
2. Targeted Therapy
Pharmacological approaches to target skin cancer have been widely studied in the last two decades. Targeting cancer by focusing on cancer type-specific genetic alterations and the most consequential signaling cascade abnormalities in tumor initiation and progression are direct and profound applications to rehabilitate cellular homeostasis.
In cutaneous melanoma, the most common druggable mutations are found in the MAPK/ERK pathway [7][8] (Figure 1b). Up to half of the patients diagnosed with CM carry activating mutations in the serine-threonine kinase of the BRAF gene (BRAF-V600), with almost a quarter of patients (15–25%) carrying mutations in the RAS gene (Q61R, Q61K), which downstream activate RAF, MEK, and ERK. [9][10][11]. Other mutations associated with poor outcomes include CDKN2A and TP53 in ~13% and ~15% of melanoma patients, respectively [12]. MAPK/ERK pathway mutations enhance proliferation, survival, and spread of melanoma cells, and thus, patients carrying the mutation are eligible for treatment with BRAF and MEK inhibitors. In BRAF-mutant metastatic melanoma, BRAF and MEK inhibitors have proven to improve survival, although half of the patients develop resistance within a year [13][14]. Moreover, treatment with BRAF and MEK inhibitors is associated with toxicity, as shown in a case of widespread acneiform rash developing 2 months after the initiation of anti-BRAF plus anti-MEK combination therapy (Figure 2). Several adverse events (AE), including hyperkeratosis, rash, alopecia, skin papilloma, palmar-plantar hyperkeratosis, and arthralgia, as well as rare adverse events such as cutaneous SCC and pyrexia were observed. Most used BRAF/MEK inhibitor combinations include Dabrafenib/Trametinib, Vermurafenib/Cobimetinib, and Encorafenib/Binimetinib. In addition, BRAFi was associated with increased antigen expression, lymphocyte homing, and a decrease in immunosuppressive cytokine release in melanoma cell lines and patients’ biopsies, providing a rationale for ICI-BRAF/MEKi combinations [15][16][17][18]. This was further explored in a first-in-human clinical trial of dabrafenib, trametinib, and pembrolizumab in which 11/15 patients (73%) showed an objective response and 6/15 continued with a response at a median follow-up of 27 months. Triple therapy had higher PFS (16 months) compared to dabrafenib and trametinib double therapy (10.3 months) with a median duration of 18.7 months. [18][19]. Recently, a randomized, double-blind phase 3 trial on 514 patients suffering from stage III-IV BRAFV600-positive melanoma evaluated the use of atezolizumab in combination with vermurafenib and cobimetinib. Progression-free survival was significantly prolonged from 10.6 months in the control group to 15.1 months in the atezolizumab group with similar ORR (65% vs. 66%) [20]. While triple therapy combinations were prone to a higher incidence of AEs, these studies were the first to indicate that BRAFi/MEKi/anti-PD1/PDL1 combinations have the potential to increase the frequency of long-lasting antitumor responses in BRAF v600-mutant melanoma patients [18][20].
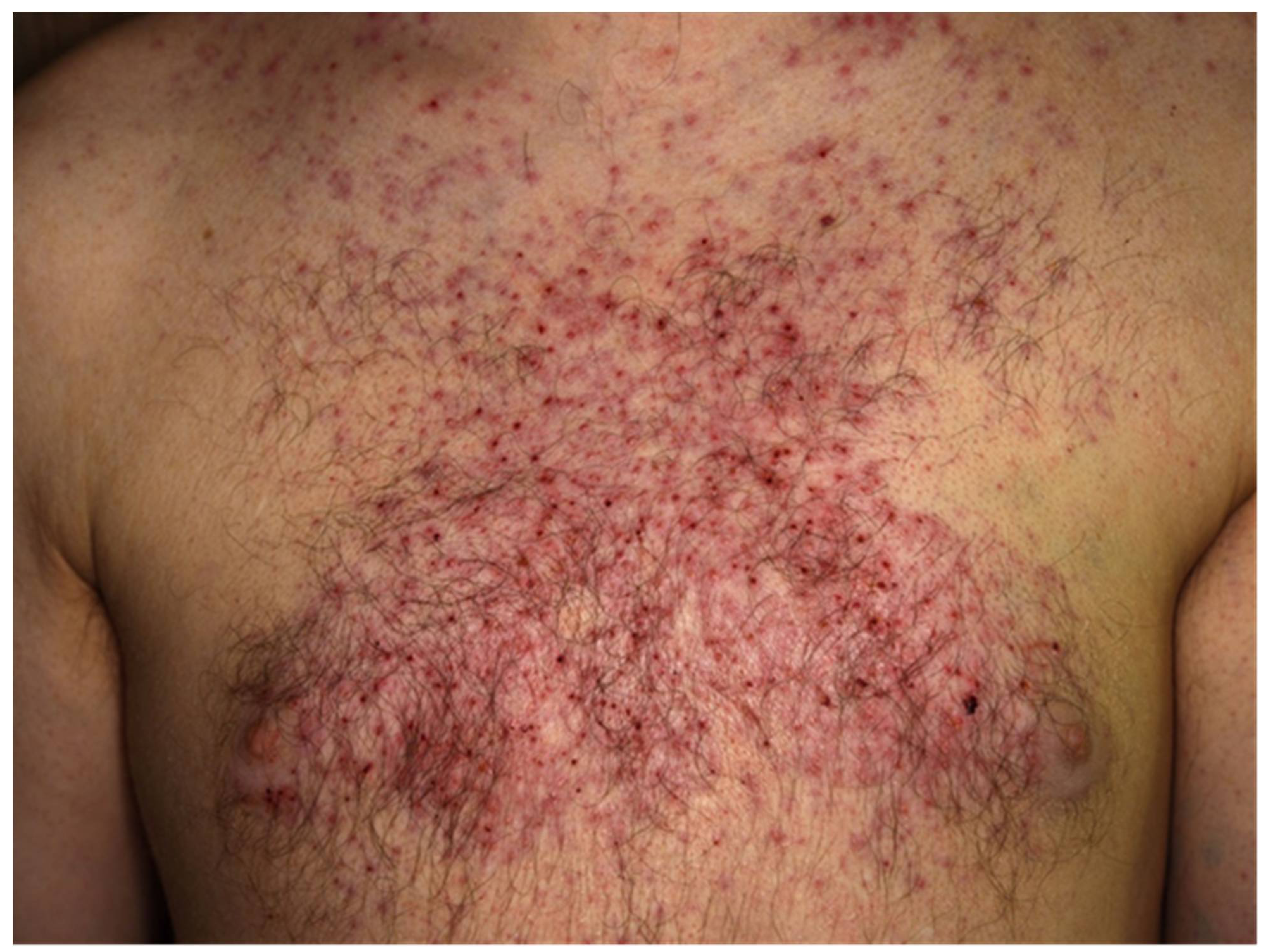
Figure 2.
Targeted therapies are also associated with AEs occurring of any grade in almost all patients treated with the combination therapy. In patients treated with BRAFi/MEKi, grade 3 to 4 AEs have been observed in about 50%. The discontinuation rate due to AEs of BRAFi/MEKi is about 15% and thus much lower as compared to combinations of anti-PD-1 and anti-CTLA-4. The most frequently reported AEs of BRAFi/MEKi include cutaneous toxicities (Figure 2; e.g., acneiform rashes, photosensitivity, palmoplantar hyperkeratosis), diarrhea, pyrexia, hepatic toxicities, arthralgia, cardiovascular toxicities (e.g., hypertension, QT-prolongation), ocular AEs (Figure 3; retinal detachment, uveitis), and rarely pneumonitis.
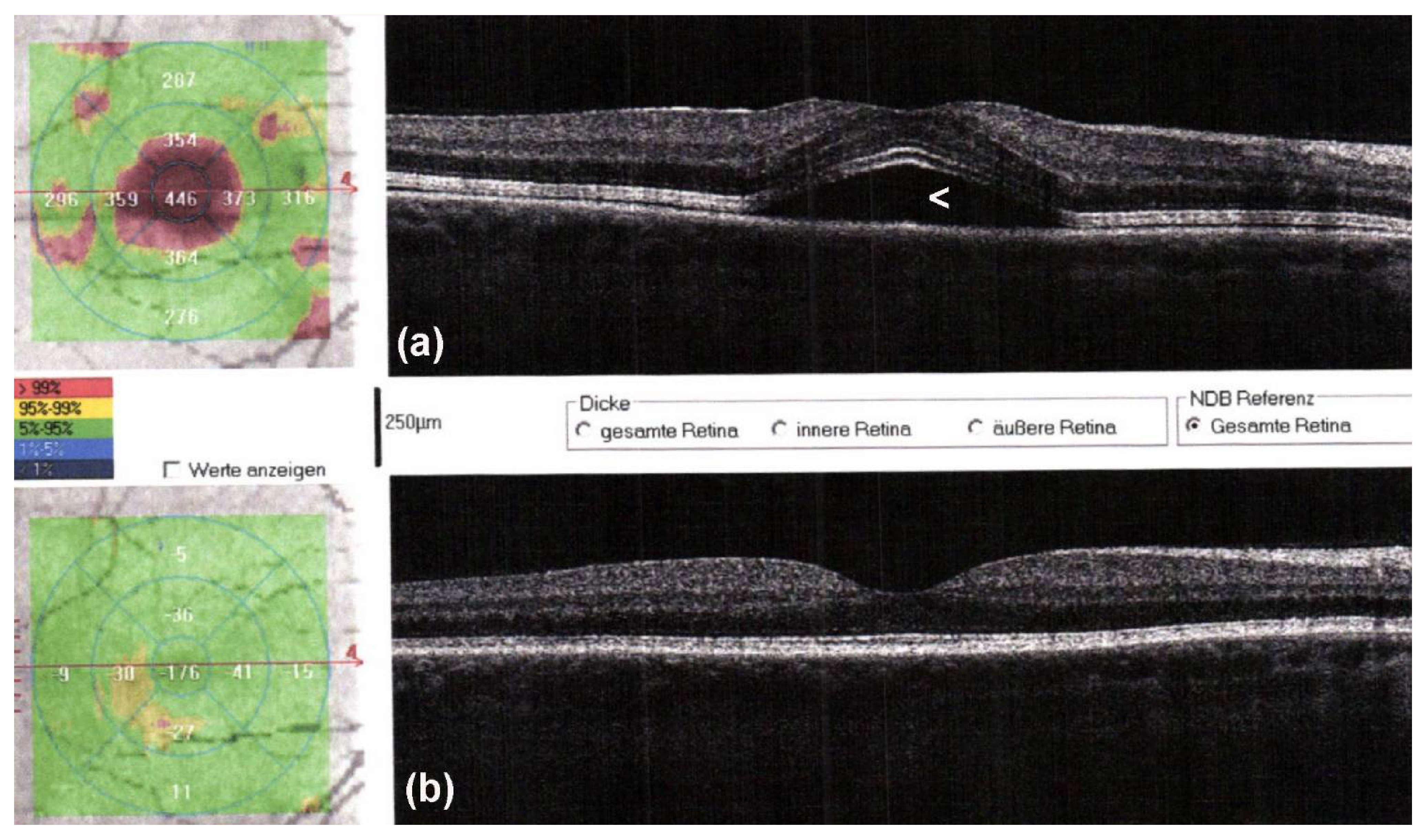
Figure 3. Optical coherence tomography (OCT) scans of a male patient with metastatic melanoma started targeted therapy with encorafenib and binimetinib. Two days after therapy initiation, he noticed blurry vision. On OCT, serous retinal detachment with subfoveolar fluid (a, arrowhead) is observed, which spontaneously resolved after withholding targeted therapy for 5 days (b).
3. Conclusions and Future Perspective
The development of patient-centered approaches for targeted treatment will be one of the main challenges in CM care in the next decade. Combining therapeutic options offers the unique opportunity to tailor treatment to patient- and cancer-specific conditions utilizing the synergistic effects of existing therapeutic approaches (Figure 1e). Combinations of several systemic treatment options such as ICI, BRAFi/MEKi, and angiogenic inhibitors for metastatic melanoma, as well as intratumoral and systemic immunotherapeutic applications to prime and induce immuno-sensitivity of locally advanced CM show promising results and potential to overcome acquired and endogenous treatment resistance. Nevertheless, limitations for increasingly complex treatment combinations remain. Patients treated with ICI can exhibit a range of non-cutaneous and cutaneous irAEs, which range in severity and can have detrimental effects on a patient’s quality of life and well-being, limiting subsequent treatment options [21]. Combining two or more therapeutic approaches, although potentially leading to improved efficacy, can increase the incidence and severity of irAEs. As discussed in previous sections, there is a range of potentially life-threatening irAEs affecting a multitude of organs, but there are also less severe cutaneous irAEs substantially impacting patient’s quality of life. Cutaneous irAEs are the most common adverse events occurring in up to 50% of patients undergoing ICI resemble autoimmune disorders mostly presenting as primary dermatoses [22][23][24]. Future investigations will have to address emerging non-cutaneous and cutaneous AEs as well as potential augmentation of treatment interventions combining different modes of action whilst minimizing toxicity. In addition, we would like to emphasize that the survival of patients with CM is significantly improved when the tumor is detected at a very early stage—a time when simple surgery is fully sufficient to cure the patient’s CM. Hence, we also want to refer to emerging novel techniques under current investigation, which might significantly improve the accuracy of early CM diagnosis. A variety of innovative optical and acoustic technology-based techniques, such as confocal laser-scanning microscopy, optical coherence tomography (OCT), photoacoustic/ultrasound/OCT, multiphoton excited fluorescence imaging, and stepwise two-photon excited fluorescence, have been developed to increase the diagnostic accuracy for the non-invasive melanoma diagnosis [25][26][27][28][29][30][31]. Advancing diagnostic tools and biomarkers to identify subgroup beneficiaries of specific treatment combinations prior to therapy can thus become the next step in patient-centered personalized CM treatment.
References
- Paulson, K.G.; Gupta, D.; Kim, T.S.; Veatch, J.R.; Byrd, D.R.; Bhatia, S.; Wojcik, K.; Chapuis, A.G.; Thompson, J.A.; Madeleine, M.M.; et al. Age-Specific Incidence of Melanoma in the United States. JAMA Dermatol. 2020, 156, 57–64.
- Apalla, Z.; Lallas, A.; Sotiriou, E.; Lazaridou, E.; Ioannides, D. Epidemiological trends in skin cancer. Dermatol. Pract. Concept. 2017, 7, 1–6.
- Guy, G.P.; Thomas, C.C.; Thompson, T.; Watson, M.; Massetti, G.M.; Richardson, L.C. Vital signs: Melanoma incidence and mortality trends and projections—United States, 1982–2030. MMWR Morb. Mortal. Wkly. Rep. 2015, 64, 591.
- Skin Cancer Foundation. Skin Cancer Facts and Statistics. Skin Cancer Information. Last Updated: January 13, 2021. Available online: (accessed on 25 April 2021).
- Seite, S.; Del Marmol, V.; Moyal, D.; Friedman, A. Public primary and secondary skin cancer prevention, perceptions and knowledge: An international cross-sectional survey. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 815–820.
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953.
- Savoia, P.; Fava, P.; Casoni, F.; Cremona, O. Targeting the ERK Signaling Pathway in Melanoma. Int. J. Mol. Sci. 2019, 20, 1483.
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102.
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180.
- Menzies, A.M.; Haydu, L.E.; Visintin, L.; Carlino, M.S.; Howle, J.R.; Thompson, J.F.; Kefford, R.F.; Scolyer, R.A.; Long, G.V. Distinguishing Clinicopathologic Features of Patients with V600E and V600K BRAF-Mutant Metastatic Melanoma. Clin. Cancer Res. 2012, 18, 3242–3249.
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954.
- Zhang, T.; Dutton-Regester, K.; Brown, K.M.; Hayward, N.K. The genomic landscape of cutaneous melanoma. Pigment. Cell Melanoma Res. 2016, 29, 266–283.
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic mela-noma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365.
- Trunzer, K.; Pavlick, A.C.; Schuchter, L.; Gonzalez, R.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; Kim, K.B.; Weber, J.S.; et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J. Clin. Oncol. 2013, 31, 1767–1774.
- Bradley, S.D.; Chen, Z.; Melendez, B.; Talukder, A.; Khalili, J.S.; Rodriguez-Cruz, T.; Liu, S.; Whittington, M.; Deng, W.; Li, F.; et al. BRAFV600E Co-opts a Conserved MHC Class I Internalization Pathway to Diminish Antigen Presentation and CD8+ T-cell Recognition of Melanoma. Cancer Immunol. Res. 2015, 3, 602–609.
- Mandalà, M.; De Logu, F.; Merelli, B.; Nassini, R.; Massi, D. Immunomodulating property of MAPK inhibitors: From translational knowledge to clinical implementation. Lab. Investig. 2016, 97, 166–175.
- Frederick, D.T.; Piris, A.; Cogdill, A.P.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF Inhibition Is Associated with Enhanced Melanoma Antigen Expression and a More Favorable Tumor Microenvironment in Patients with Metastatic Melanoma. Clin. Cancer Res. 2013, 19, 1225–1231.
- Ribas, A.; Lawrence, D.; Atkinson, V.; Agarwal, S.; Miller, W.H.; Carlino, M.S.; Fisher, R.; Long, G.V.; Hodi, F.S.; Tsoi, J.; et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat. Med. 2019, 25, 936–940.
- Ascierto, P.A.; Ferrucci, P.F.; Fisher, R.; Del Vecchio, M.; Atkinson, V.; Schmidt, H.; Schachter, J.; Queirolo, P.; Long, G.V.; Di Giacomo, A.M.; et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat. Med. 2019, 25, 941–946.
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAFV600 mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844.
- Michot, J.; Bigenwald, C.; Champiat, S.; Collins, M.; Carbonnel, F.; Postel-Vinay, S.; Berdelou, A.; Varga, A.; Bahleda, R.; Hollebecque, A.; et al. Immune-related adverse events with immune checkpoint blockade: A comprehensive review. Eur. J. Cancer 2016, 54, 139–148.
- Tattersall, I.W.; Leventhal, J.S. Cutaneous toxicities of immune checkpoint inhibitors: The role of the dermatologist. Yale J. Biol. Med. 2020, 93, 123.
- Habre, M.M.; Habre, S.B.S.; Kourie, H.R. Dermatologic adverse events of checkpoint inhibitors: What an oncologist should know. Immunotherapy 2016, 8, 1437–1446.
- Sibaud, V. Dermatologic Reactions to Immune Checkpoint Inhibitors: Skin Toxicities and Immunotherapy. Am. J. Clin. Dermatol. 2018, 19, 345–361.
- Gambichler, T.; Schmid-Wendtner, M.H.; Plura, I.; Kampilafkos, P.; Stücker, M.; Berking, C.; Maier, T. A multicentre pilot study investigating high-definition optical coherence tomography in the differentiation of cutaneous melanoma and melanocytic naevi. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 537–541.
- Ferrante di Ruffano, L.; Dinnes, J.; Deeks, J.J.; Chuchu, N.; Bayliss, S.E.; Davenport, C.; Takwoingi, Y.; Godfrey, K.; O’Sullivan, C.; Matin, R.N.; et al. Optical coherence tomography for diagnosing skin cancer in adults. Cochrane Database Syst. Rev. 2018, 4, CD013189.
- Shahriari, N.; Grant-Kels, J.M.; Rabinovitz, H.; Oliviero, M.; Scope, A. Reflectance confocal microscopy: Diagnostic criteria of common benign and malignant neoplasms, dermoscopic and histopathologic correlates of key confocal criteria, and diagnostic algorithms. J. Am. Acad. Dermatol. 2021, 84, 17–31.
- Turani, Z.; Fatemizadeh, E.; Blumetti, T.; Daveluy, S.; Moraes, A.F.; Chen, W.; Mehregan, D.; Andersen, P.E.; Nasiriavanaki, M. Optical Radiomic Signatures Derived from Optical Coherence Tomography Images Improve Identification of Melanoma. Cancer Res. 2019, 79, 2021–2030.
- Kratkiewicz, K.; Manwar, R.; Rajabi-Estarabadi, A.; Fakhoury, J.; Meiliute, J.; Daveluy, S.; Mehregan, D.; Avanaki, K.M. Photoacoustic/Ultrasound/Optical Coherence Tomography Evaluation of Melanoma Lesion and Healthy Skin in a Swine Model. Sensors 2019, 24, 2815.
- Hai, P.; Qu, Y.; Li, Y.; Zhu, L.; Shmuylovich, L.; Cornelius, L.A.; Wang, L.V. Label-free high-throughput photoacoustic tomography of suspected circulating melanoma tumor cells in patients in vivo. J. Biomed. Opt. 2020, 25, 1–17.
- Meng, X.; Chen, J.; Zhang, Z.; Li, K.; Li, J.; Yu, Z.; Zhang, Y. Non-invasive optical methods for melanoma diagnosis. Photodiagnosis Photodyn. Ther. 2021, 34, 102266.