The heterotrimeric Sec61 complex of the ER membrane represents the major entry point for precursor polypeptides into the membrane or lumen of the ER. It foreprmesents a polypeptide-conducting channel, who's gating (i.e. opening and closing) involves various interactions partners. Mutations in the genes, which are coding for the Sec61 subunits or their interaction partners, can cause diseases (termed Sec61-channelopathies).
- BiP
- common variable immunodeficiency
- congenital disorder of glycosylation
- endoplasmic reticulum
- neutropenia
- polycystic liver disease
- Sec61-channelopathies
- Sec62
- Sec63
- SSR/TRAP complex
1. Introduction
The heterotrimeric Sec61 complex of the ER membrane represents the major entry point for precursor polypeptides into the membrane or lumen of the ER (Figure 1 and Figure 2) [1][2][3][4][5][6][7][8][9][10][11][12][13]. Therefore, it can form an aqueous polypeptide-conducting channel, which also allows the passage of Ca2+ in the opposite direction [14][15][16][17][18][19][20][21][22][23]. The channel exists in two conformations, an open and a closed state that are in a dynamic equilibrium with each other (Figure 2). The closed conformation is impermeable even to Ca2+. Thus, the Sec61 complex is a precursor-gated channel, which operates either coupled to translation (in co-translational transport) or after completion of translation (in post-translational transport). Sec61 gating to the open state is not solely facilitated by its substrates, the amino-terminal signal peptides (SPs) or transmembrane helices (TMHs) of precursor polypeptides [24][25][26][27][28][29], it is also supported by ribosomes in co-translational transport [6][9] and/or by several Sec61 interaction partners, such as translocon-associated protein or TRAP complex [7][10][30][31] and Sec62/Sec63 complex in cooperation with BiP [23][32][33][34][35], respectively. Here, the latter are defined as allosteric effectors of the channel since they interact with the complex at sites, which are distinct from the precursor binding sites. Channel closing also appears to be facilitated by allosteric effectors, such as the ER-lumenal BiP [23] and/or the cytosolic Ca2+-calmodulin (CaM) [22]. In our opinion, Sec61 channel gating can best be described in analogy to an enzyme-catalyzed reaction (Figure 3): Channel opening and closing represent two energetically un-favorable reversible reactions and the substrates and effectors are the catalysts, which lower the activation energy for the required conformational transitions by binding to the Sec61 complex [11][12].
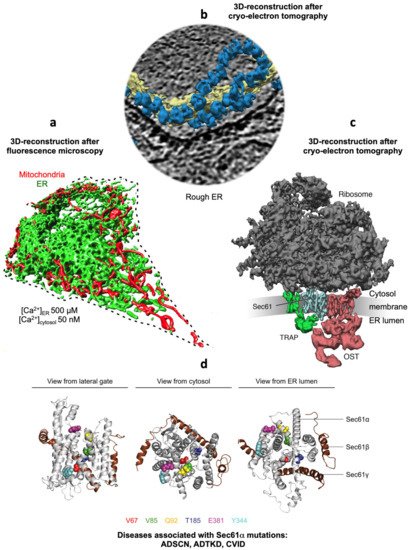
Figure 1. Collage of 3D reconstructions of a nucleated mammalian cell, a section of rough ER in such a cell and a ribosome-bound Sec61 translocon. (a) Represents a 3D reconstruction after live cell fluorescence imaging, following import of GFP into the ER and of RFP into the mitochondria. The plasma membrane is indicated by a dashed line. Typical concentrations of free Ca2+ are given for cytosol and ER of a resting cell. (b) depicts a 3D reconstruction of cellular rough ER after CET of a slice through the respective tomogram. ER membranes are shown in yellow; 80S ribosomes are shown in blue. (c) represents a 3D reconstruction of the native ribosome-translocon complex in rough microsomes. Here, the membrane density was removed to highlight membrane integral parts of the translocon complex. TMHs for Sec61 complex, TRAP and OST can be distinguished [10]. Helix 51 of an rRNA expansion segment and ribosomal protein eL38 represent the contact sites of TRAPγ, but are hidden by other ribosomal densities. (d) shows the PDB 3j7q structures for the Sec61 channel as seen from the indicated positions; disease associated point mutations are indicated. The collage is based on [11][12]. See text for details.
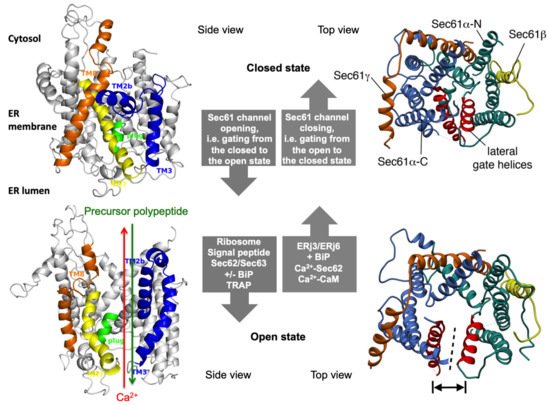
Figure 2. The concept of gating of the heterotrimeric Sec61 complex by signal peptides and allosteric effectors. The Sec61 channel is shown in its modeled closed (top) and open (bottom) conformational states, as viewed from the plane of the membrane (left) and in atomic models (PDB 3j7q, PDB 3jc2) as seen from the cytosol (right), respectively [11][12]. These two states are proposed to be in a dynamic equilibrium with each other. The fully open state of the Sec61 channel allows the initial entry of precursor polypeptides from the cytosol into the ER lumen and ER membrane, respectively, and is experimentally observed as cleavage of signal peptides by signal peptidase on the luminal side of the ER membrane. In addition, it allows the passive efflux of Ca2+ from the ER lumen into the cytosol and is visible in live cell Ca2+ imaging in cytosol and ER lumen. Ca2+ efflux may also be possible in the expected transition state (not shown), which may be identical to the so-called primed state that can be induced by ribosomes in co-translational- and by the Sec62/Sec63 complex in post-translational-transport. The conformational changes of the modelled Sec61 complex were previously morphed and the role of BiP plus an ERj co-chaperone, such as Sec63 and ERj1, respectively, visualized for co-translational transport at (see Data Availability). The Ca2+-permeability of the open Sec61 channel as observed by live cell Ca2+ imaging can be seen in the video file (see Data Availability).
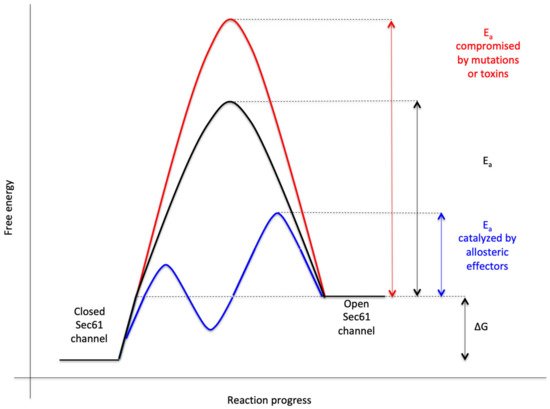
Figure 3. Energetics and kinetics of Sec61 channel gating. In our view, the TRAP− or Sec62/Sec63 +/− BiP-mediated Sec61 channel gating is best considered in analogy to an enzyme-catalysed reaction. Accordingly, TRAP, Sec62, Sec63 or BiP reduce the energetic barrier for full channel opening, which can apparently be reinforced by Sec61 channel inhibitors, such as cyclic heptadepsipeptides (such as CAM741) or certain eeyarestatins (such as ES24). At least in the case of ES24, binding of the inhibitor within the channel pore arrests the channel in a partially open state (termed “foot in the door”), which maybe identical with the primed state and is compatible with Ca2+-efflux but not with full channel opening for protein translocation. TRAP and BiP contribute to full channel opening by direct interaction with ER luminal loops 5 and 7, respectively, of Sec61α (see below). SEC61A1 mutations can increase the energy barrier for channel opening per se (V67G, V85D, and Q92R mutation) or indirectly, such as by interfering with BiP binding (Y344H mutation). Notably, all these effects are precursor specific because the amino-terminal SPs are either efficient or inefficient in driving Sec61 channel opening. Typical for an enzyme-catalysed reaction, BiP can also support efficient gating of the Sec61 channel to the closed state, i.e., the reverse reaction.
The Hsp70-type molecular chaperone immunoglobulin heavy chain binding protein or BiP [36] does not only support Sec61 channel opening for ER protein import [23][32], but also can bind to incoming precursor polypeptides and act on these as a molecular ratchet [37]. Thus, typical for an Hsp70, the ATP- and Ca2+-dependent BiP modulates the conformation of a folded protein complex, the Sec61 channel, plus interacts with a more or less unfolded polypeptide chain as it emerges from the Sec61 channel, thereby contributing to a unidirectional or irreversible transport process. Also typical for an Hsp70, both BiP activities involve an ATPase cycle and their own allosteric effectors, i.e., J-domain-proteins (JDPs) [38] or Hsp40-type co-chaperones, termed ERj- or ERdj-proteins, and nucleotide exchange factors (NEFs). Following the same principles and interactions, BiP also plays a central role in folding and assembly of newly-imported polypeptides, such as heavy and light chains of immunoglobulins in the plasma cells of the immune system [39], and supports efficient Sec61 channel closing to preserve Ca2+ homeostasis [23].
2. The Human Sec61 Translocon
Protein import into the ER is the first step in the biogenesis of precursors of about 10,000 different soluble and membrane proteins of nucleated human cells, which amounts to about 30% of the proteome [40][41][42]. All these proteins fulfill their functions either in the membrane or lumen of the ER (plus the connected nuclear envelope), in one of the organelles of the pathways for endo- and exocytosis (i.e., ERGIC, Golgi apparatus, endosome, lysosome), in lipid droplets or at the cell surface as plasma membrane- or secretory-proteins. ER protein import involves the two stages of membrane targeting and insertion of nascent membrane proteins into or translocation of soluble precursor polypeptides across the ER membrane. Typically, both processes depend on SPs or TMHs at the amino-termini of the precursor polypeptides [24][25][26][27][28][29]. In general, these SPs have a tripartite structure. They comprise a more or less positively charged amino-terminal or N-region, a central hydrophobic or H-region and a slightly polar carboxy-terminal or C-region (Figure 4). Other than that, they do not have sequence homologies and, as a matter of fact, show quite some variability with respect to length (15–50 amino acid residues) as well as overall properties (see below). Interestingly, various human hereditary diseases are the result of single point mutations in the SPs of certain precursor polypeptides (such as preproinsulin and preprorenin), which result in failure of these SPs to deliver their otherwise functional mature forms to the correct cellular location, thus, highlighting the fact that these amino-terminal SPs were fine-tuned to their respective receptors by evolution [43][44][45]. In addition, insertion of SPs may occur co- or post-translationally and are facilitated by various pathways and components, which reside in the cytosol and the ER membrane or lumen, respectively (Table 1).
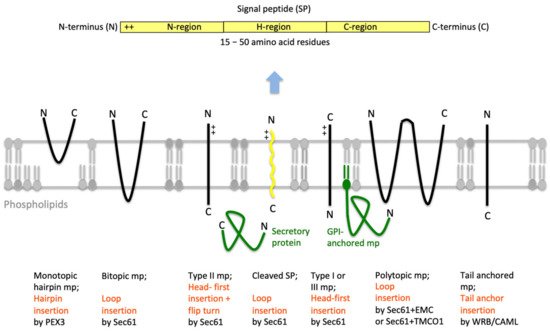
Figure 4. Features of amino-terminal signal peptides and ER membrane proteins. The cartoon depicts the signal peptides (SPs, shown in yellow) and six types of ER membrane proteins (in black), together with their membrane protein type and mechanism of membrane insertion (both indicated below the cartoon). Cleavable SPs (in yellow) can facilitate ER import of secretory proteins (in green), glycosylphosphatidylinositol (GPI)-anchored membrane proteins (in green) and various types of membrane proteins, except for hairpin-, type II- and tail anchored-membrane proteins. Positively charged amino acid residues (+) play an important role in membrane protein and SP orientation, i.e. typically, follow the positive inside rule. Bitopic and polytopic proteins can also involve SPs and have the opposite instead of the shown orientation. Alternatively, amino-terminal transmembrane helices (TMHs), which serve as SPs, facilitate membrane insertion. The shown bitopic protein is also named double-spanning membrane protein, the example polytopic protein is also named tetra-spanning membrane protein, if the shown type I membrane protein did not involve a cleavable SP it is also defined as signal anchor protein. In the case of membrane proteins with amino-terminal TMHs, membrane insertion typically involves the same components and mechanisms, which deliver secretory proteins (in green) and glycosylphosphatidylinositol (GPI)-anchored membrane proteins (in green) to the ER lumen. In certain cases, however, auxiliary membrane protein insertases, such as EMC or TMCO1 complex play a role. The latter two membrane protein complexes can also operate as stand-alone membrane protein insertases, an activity they have in common with the PEX- and the TRC-systems. Following their ER import, GPI-anchored membrane proteins become membrane anchored via their carboxy-termini by GPI-attachment. C, carboxy-terminus; N, amino-terminus.
3. Gating of the Sec61 Channel by BiP
The Hsp70-type molecular chaperone immunoglobulin heavy chain binding protein or BiP, which was discovered by I. Haas, also goes through a cycle of open and closed conformation [36][46][47][48][49][50][51][52][53][54][55][56][57]. However, in this case the description refers to the state of the substrate-binding domain (SBD). It also involves allosteric effectors in its conformational changes and, in contrast to Sec61, the hydrolysis of ATP. It does not only support Sec61 channel opening for ER protein import [23][32][58][59][60], but also can bind to and act on the incoming precursor polypeptide as a molecular ratchet [37]. Thus, typical for an Hsp70, the ATP- and Ca2+-dependent BiP modulates the conformation of a folded protein complex, the Sec61 channel, plus interacts with a more or less unfolded polypeptide chain as it emerges from the Sec61 channel, thereby vectorizing the transport process. These two kinds of substrates (folded and unfolded substrates) have previously also been observed for various other Hsp70s, such as sigma factor 32 in the bacterial cytosol and clathrin triscelions in the cytosol of human cells. Also typical for an Hsp70, both BiP activities involve an ATPase cycle, where the chaperone goes through states of substrate trapping in its ADP-bound state and substrate release in its ATP-bound state (Figure 5b). Furthermore, these activities involve JDPs, termed ERj- or ERdj-proteins, which stimulate the hydrolysis of BiP-bound ATP [38][61], and NEFs [62][63][64][65][66][67][68], which stimulate the exchange of ADP for ATP, thereby affecting BiP conformations allosterically [54][55]. Following the same principles and interactions, BiP also plays a central role in folding and assembly of newly-imported polypeptides, such as heavy and light chains of immunoglobulins in plasma cells of the immune system [39][46][47], and supports efficient Sec61 channel closing to preserve Ca2+ homeostasis [23][69]. In addition, BiP is a key player in various Ca2+-dependent and -independent signal transduction pathways, which report on ER energy homeostasis and proteostasis, respectively, and can first increase ATP/ADP exchange between ER and cytosol, next increase the folding- and ERAD-capacity via UPR and, at last, switch to apoptosis under conditions of ever increasing protein mis-folding or ER stress [70][71][72][73][74][75][76][77][78][79][80].

Figure 5. Topology and functionally relevant domains of the heterotrimeric Sec61 complex and its allosteric effectors TRAP, BiP, Sec62 and Sec63. The membrane topology of the three subunits of the mammalian Sec61 complex indicates binding sites (termed BS) for the ribosome (R, cytosolic loops 6 and 8 of Sec61a) and for Ca2+-calmodulin (CaM, IQ) and BiP (loop 7 of Sec61a). The relevant allosteric effectors of BiP (ERjs and NEFs) are also shown in, as are additional interaction partners of Sec62 (LC3) and Sec63 (nucleoredoxin or NRX, calumenin or Calu). Recent work demonstrated that IRE1a interacts with Sec61 and connects protein translocation and Ca2+ leakage with UPR. Furthermore, functional domains (J-domain, NBD, SBD, TPR) plus motifs (IQ, EF hand) and disease-associated mutations of Sec61a are indicated (in red; amino acid residues are given in single letter code). C, carboxy-terminus; N, amino-terminus. Notably, recent 3D reconstructions after single particle cryo-electron microscopic analysis of the yeast SEC complex that comprises Sec62p, Sec63p, Sec71p and Sec72p showed that in the post-translationally acting Sec61 complex, the Sec62/Sec63 sub-complex interacts with the cytosolic loops 6 and 8 on the cytosolic face of the Sec61 complex, and that the ER luminal domain of Sec63 interacts with ER luminal loop 5 of Sec61a.
4. Sec61-Channelopathies
As outlined above, the Sec61 channel of the human ER membrane and its allosteric effectors (TRAP, Sec62, Sec63, and BiP) plus BiP´s co-chaperones and NEFs play central roles in extra- and intra-cellular proteostasis as well as in intracellular Ca2+-homeostasis. Therefore, the term Sec61-channelopathies was coined for diseases, which are the result of toxin-driven or hereditary defects in one of the three Sec61 subunits themselves or in one of the many allosteric effectors of the Sec61 channel (Figure 6) [81]. In general, genetically-determined defects can affect a single or both alleles coding for a certain component and are termed heterozygous or homozygous; heterozygous mutations can result in haploinsufficiency, where the product of the wild type allele cannot compensate the loss of function of the mutated one, or in a dominant negative effect of the product of the mutated allele. Bacterial and fungal toxins can have similar effects as mutations [82]. On the other hand, some components of the interaction network of our interest here appear to have overlapping functions and, therefore, may be partially able to substitute for each other. In addition, some of the components were found to be overproduced in various types of tumor diseases, suggesting that overproduction and gain-of-function of a component can cause or support a disease state, too. Next, we summarize the current knowledge about these diseases, knowing the basic medical principle that “he who has fleas can also have lice” or, at the molecular level, that a certain disease may be the result of simultaneous lack of one function of the component and dominant negative effect of one of its additional functions.
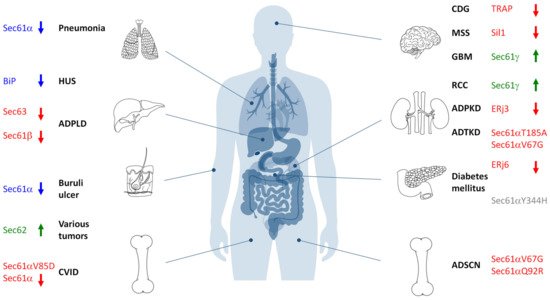
Figure 6. Hereditary and acquired diseases that are linked to the Sec61 complex and its allosteric effectors. The figure highlights various disease phenotypes, which are discussed in the text. Proteins affected in human hereditary diseases are indicated in red, protein targets of toxin-determined human diseases in blue, over-produced proteins in human tumor diseases in green (see below) and a genetically-determined variant causing murine Diabetes mellitus in grey. The arrows point upwards for increased activity of the indicated component and downwards for decreased activity. ADPKD, autosomal dominant polycystic kidney disease; ADPLD, autosomal dominant polycystic liver disease; ADSCN, autosomal dominant severe congenital neutropenia; ADTKD, autosomal dominant tubulo-interstitial kidney disease; CDG, congenital disorder of glycosylation; CVID, common variable immunodeficiency; GBM, glioblastoma multiforme; HUS, hemolytic uremic syndrome; MSS, Marinesco–Sjögren syndrome; RCC, renal cell carcinoma.
It is noteworthy in the context of Sec61-channelopathies that endosome-resident Sec61 complexes were shown to be involved in antigen transport from endosomes into the cytosol for cross-presentation to CD8+ T cells [83]. In the so-called endosome to cytosol pathway of cross-presentation, antigens are exported from endosomes into the cytosol by endosome-resident Sec61 channels and degraded by the proteasome, in possible analogy to Sec61-dependent ERAD. Therefore, this moonlighting function of the Sec61 channel has always to be considered in Sec61-related diseases, as may equally be true for possible specialized functions of Sec61 channels in ERAD.
References
- Simon, S.M.; Blobel, G. A protein-conducting channel in the endoplasmic reticulum. Cell 1991, 65, 371–380.
- Görlich, D.; Prehn, S.; Hartmann, E.; Kalies, K.-U.; Rapoport, T.A. A mammalian homolog of SEC61p and SECYp is associated with ribosomes and nascent polypeptides during translocation. Cell 1992, 71, 489–503.
- Görlich, D.; Rapoport, T.A. Protein translocation into proteoliposomes reconstituted from purified components of the endoplasmic reticulum membrane. Cell 1993, 75, 615–630.
- Hartmann, E.; Sommer, T.; Prehn, S.; Görlich, D.; Jentsch, S.; Rapoport, T.A. Evolutionary conservation of components of the protein translocation complex. Nature 1994, 367, 654–657.
- Pfeffer, S.; Brandt, F.; Hrabe, T.; Lang, S.; Eibauer, M.; Zimmermann, R.; Förster, F. Structure and 3D arrangement of ER-membrane associated ribosomes. Structure 2012, 20, 1508–1518.
- Voorhees, R.M.; Fernández, I.S.; Scheres, S.H.W.; Hegde, R.S. Structure of the mammalian ribosome-Sec61 complex to 3.4 Å resolution. Cell 2014, 157, 1632–1643.
- Pfeffer, S.; Dudek, J.; Gogala, M.; Schorr, S.; Linxweiler, J.; Lang, S.; Becker, T.; Beckmann, R.; Zimmermann, R.; Förster, F. Structure of the mammalian oligosaccharyltransferase in the native ER protein translocon. Nat. Commun. 2014, 5, 3072.
- Pfeffer, S.; Burbaum, L.; Unverdorben, P.; Pech, M.; Chen, Y.; Zimmermann, R.; Beckmann, R.; Förster, F. Structure of the native Sec61 protein-conducting channel. Nat. Commun. 2015, 6, 8403.
- Voorhees, R.M.; Hegde, R.S. Structure of the Sec61 channel opened by a signal peptide. Science 2016, 351, 88–91.
- Pfeffer, S.; Dudek, J.; Ng, B.; Schaffa, M.; Albert, S.; Plitzko, J.; Baumeister, W.; Zimmermann, R.; Freeze, H.; Engel, B.D.; et al. Dissecting the molecular organization of the translocon-associatecd protein complex. Nat. Commun. 2017, 8, 14516.
- Lang, S.; Pfeffer, S.; Lee, P.-H.; Cavalié, A.; Helms, V.; Förster, F.; Zimmermann, R. An update on Sec61 channel function, mechanisms, and related diseases. Front. Physiol. 2017, 8, 887.
- Lang, S.; Nguyen, D.; Pfeffer, S.; Förster, F.; Helms, V.; Zimmermann, R. Current state of affairs on the eukaryotic ribosome-translocon complex, in Macromolecular Complexes II: Structure and Function. Subcell. Biochem. 2019, 93, 83–141.
- Gemmer, M.; Förster, F. A clearer picture of the ER translocon complex. J. Cell Sci. 2020, 133, jcs231340.
- Lomax, R.B.; Camello, C.; Van Coppenolle, F.; Petersen, O.H.; Tepikin, A.V. Basal and physiological Ca2+ leak from the endoplasmic reticulum of pancreatic acinar cells. Second messenger-activated channels and translocons. J. Biol. Chem. 2002, 277, 26479–26485.
- Wirth, A.; Jung, M.; Bies, C.; Frien, M.; Tyedmers, J.; Zimmermann, R.; Wagner, R. The Sec61p complex is a dynamic precursor activated channel. Mol. Cell 2003, 12, 261–268.
- Van Coppenolle, F.; Vanden Abeele, F.; Slomianny, C.; Flourakis, M.; Hesketh, J.; Dewailly, E.; Prevarskaya, N. Ribosome-translocon complex mediates calcium leakage from endoplasmic reticulum stores. J. Cell Sci. 2004, 117, 4135–4142.
- Zhang, S.L.; Yeromin, A.V.; Zhang, X.H.; Yu, Y.; Safrina, O.; Penna, A.; Roos, J.; Stauderman, K.A.; Cahalan, M.D. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc. Natl. Acad. Sci. USA 2006, 103, 9357–9362.
- Flourakis, M.; Van Coppenolle, F.; Lehen’kyi, V.; Beck, B.; Skryma, R. Passive calcium leak via translocon is a first step for iPLA2-pathway regulated store operated channels activation. FASEB J. 2006, 20, 1215–1217.
- Giunti, R.; Gamberucci, A.; Fulceri, R.; Banhegyi, G. Both translocon and a cation channel are involved in the passive Ca2+ leak from the endoplasmic reticulum: A mechanistic study on rat liver microsomes. Arch. Biochem. Biophys. 2007, 462, 115–121.
- Ong, H.L.; Liu, X.; Sharma, A.; Hegde, R.S.; Ambudkar, I.S. Intracellular Ca2+ release via the ER translocon activates store-operated calcium entry. Pflug. Arch. 2007, 453, 797–808.
- Lang, S.; Erdmann, F.; Jung, M.; Wagner, R.; Cavalié, A.; Zimmermann, R. Sec61 complexes form ubiquitous ER Ca2+ leak channels. Channels 2011, 5, 228–235.
- Erdmann, F.; Schäuble, N.; Lang, S.; Jung, M.; Honigmann, A.; Ahmad, M.; Dudek, J.; Benedix, J.; Harsman, A.; Kopp, A.; et al. Interaction of calmodulin with Sec61α limits Ca2+ leakage from the endoplasmic reticulum. EMBO J. 2011, 30, 17–31.
- Schäuble, N.; Lang, S.; Jung, M.; Cappel, S.; Schorr, S.; Ulucan, Ö.; Linxweiler, J.; Dudek, J.; Blum, R.; Helms, V.; et al. BiP-mediated closing of the Sec61 channel limits Ca2+ leakage from the ER. EMBO J. 2012, 31, 3282–3296.
- Von Heijne, G. Signal sequences. J. Mol. Biol. 1985, 184, 99–105.
- Von Heijne, G. Towards a comparative anatomy of N-terminal topogenic protein sequences. J. Mol. Biol. 1986, 189, 239–242.
- Von Heijne, G.; Gavel, Y. Topogenic signals in integral membrane proteins. Eur. J. Biochem. 1988, 174, 671–678.
- Ng, D.T.; Brown, J.D.; Walter, P. Signal sequences specify the targeting route to the endoplasmic reticulum membrane. J. Cell Biol. 1996, 134, 269–278.
- Hegde, R.S.; Bernstein, H. The surprising complexity of signal peptides. Trends Biochem. Science 2006, 31, 563–571.
- Armenteros, J.J.; Salvatore, M.; Emanuelsson, O.; Winther, O.; von Heijne, G.; Elofsson, A.; Nielsen, H. Detecting sequence signals in targting peptides using deep learning. Life Sci. Alliance 2019, 2, e201900429.
- Wiedmann, M.; Kurzchalia, T.V.; Hartmann, E.; Rapoport, T.A. A signal sequence receptor in the endoplasmic reticulum membrane. Nature 1987, 328, 830–833.
- Menetret, J.F.; Hegde, R.S.; Aguiar, M.; Gygi, S.P.; Park, E.; Rapoport, T.A.; Akey, C.W. Single copies of Sec61 and TRAP associate with a nontranslating mammalian ribosome. Structure 2008, 16, 1126–1137.
- Dierks, T.; Volkmer, J.; Schlenstedt, G.; Jung, C.; Sandholzer, U.; Zachmann, K.; Schlotterhose, P.; Neifer, K.; Schmidt, B.; Zimmermann, R. A microsomal ATP-binding protein involved in efficient protein transport into the mammalian endoplasmic reticulum. EMBO J. 1996, 15, 6931–6942.
- Skowronek, M.H.; Rotter, M.; Haas, I.G. Molecular characterization of a novel mammalian DnaJ-like Sec63p homolog. Biol. Chem. 1999, 380, 1133–1138.
- Mayer, H.-A.; Grau, H.; Kraft, R.; Prehn, S.; Kalies, K.-U.; Hartmann, E. Mammalian Sec61 is associated with Sec62 and Sec63. J. Biol. Chem. 2000, 275, 14550–14557.
- Tyedmers, J.; Lerner, M.; Bies, C.; Dudek, J.; Skowronek, M.H.; Haas, I.G.; Heim, N.; Nastainczyk, W.; Volkmer, J.; Zimmermann, R. Homologs of the yeast Sec complex subunits Sec62p and Sec63p are abundant proteins in dog pancreas microsomes. Proc. Natl. Acad. Sci. USA 2000, 97, 7214–7219.
- Haas, I.G.; Wabl, M. Immunoglobulin heavy chain binding protein. Nature 1983, 306, 387–389.
- Tyedmers, J.; Lerner, M.; Wiedmann, M.; Volkmer, J.; Zimmermann, R. Polypeptide chain binding proteins mediate completion of cotranslational protein translocation into the mammalian endoplasmic reticulum. EMBO Rep. 2005, 4, 505–510.
- Kampinga, H.H.; Andreasson, C.; Barducci, A.; Cheetham, M.; Cyr, D.; Emanuelsson, C.; Genevaux, P.; Gestwicki, J.; Goloubinoff, P.; Huerta-Cepas, J.; et al. Function, evolution and structure of J-domain proteins. Cell Stress Chaperones 2018, 24, 7–15.
- Feige, M.J.; Hendershot, L.M.; Buchner, J. How antibodies fold. Trends Biochem. Sci. 2010, 35, 189–198.
- Palade, G. Intracellular aspects of protein synthesis. Science 1975, 189, 347–358.
- Blobel, G.; Dobberstein, B. Transfer of proteins across membranes: I. Presence of proteolytically processed and unprocessed nascent immunoglobulin light chains on membrane-bound ribosomes of murine myeloma. J. Cell Biol. 1975, 67, 835–851.
- Blobel, G.; Dobberstein, B. Transfer of proteins across membranes: II. Reconstitution of functional rough microsomes from heterologous components. J. Cell Biol. 1975, 67, 852–862.
- Jarjanazi, H.; Savas, S.; Pabalan, N.; Dennis, J.W.; Ozcelik, H. Biological implications of SNPs in signal peptide domains of human proteins. Proteins 2008, 70, 394–403.
- Guo, H.; Xiong, Y.; Witkowski, P.; Cui, J.; Wang, L.-J.; Sun, J.; Lara-Lemus, R.; Haataja, L.; Hutchison, K.; Shan, S.O.; et al. Inefficient translocation of preproinsulin contributes to pancreatic ß cell failure and late-onset Diabetes. J. Biol. Chem. 2014, 289, 16290–16302.
- Živná, M.; Hulkova, H.; Matignon, M.; Hodanova, K.; Vylet´al, P.; Kalbacova, M.; Baresova, V.; Sikora, J.; Blazkova, H.; Zivny, J.; et al. Dominant renin gene mutations associated with early-onset hyperuricemia, anemia, anch chronic kidney failure. Am. J. Hum. Genet. 2009, 85, 204–213.
- Bole, D.G.; Hendershot, L.M.; Kearney, J.F. Posttranslational association of immunoglobulin heavy chain binding protein with nascent heavy chains in nonsecreting and secreting hybridomas. J. Cell Biol. 1986, 102, 1558–1566.
- Munro, S.; Pelham, H.R.B. An Hsp70-like protein in the ER: Identity with the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell 1986, 46, 291–300.
- Lievremont, J.P.; Rizzuto, R.; Hendershot, L.M.; Meldolesi, J. BiP, a major chaperone protein of the endoplasmic reticulum lumen, plays a direct and important role in the storage of the rapidly exchanging pool of Ca2+. J. Biol. Chem. 1997, 272, 30873–30879.
- Tatu, U.; Helenius, A. Interactions between newly synthesized glycoproteins, calnexin and a network of resident chaperones in the endoplasmic reticulum. J. Cell Biol. 1997, 136, 555–565.
- Meunier, L.; Usherwood, Y.-K.; Chung, K.T.; Hendershot, L.M. A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol. Biol. Cell 2002, 13, 4456–4469.
- Luo, S.; Mao, C.; Lee, B.; Lee, A.S. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol. Cell Biol. 2006, 15, 5688–5697.
- Mimura, N.; Hamada, H.; Kashio, M.; Jin, H.; Toyama, Y.; Kimura, K.; Iida, M.; Goto, S.; Saisho, H.; Toshimori, K.; et al. Aberrant quality control in the endoplasmic reticulum impairs the biosynthesis of pulmonary surfactant in mice expressing mutant BiP. Cell Death Differ. 2007, 14, 1475–1485.
- Awad, W.; Estrada, I.; Shen, Y.; Hendershot, L.M. BiP mutants that are unable to interact with endoplasmic reticulum DnaJ proteins provide insights into interdomain interactions in BiP. Proc. Natl. Acad. Sci. USA 2008, 105, 1164–1169.
- Zhuravieva, A.; Gierasch, L. Substrate-binding domain conformational dynamics mediate Hsp70 allostery. Proc. Natl. Acad. Sci. USA 2015, 112, E2865–E2873.
- Kityk, R.; Vogel, M.; Schlecht, R.; Bukau, B.; Mayer, M.P. Pathways of allosteric regulation in Hsp70 chaperones. Nat. Commun. 2015, 6, 8308.
- Xu, M.; Marsh, H.M.; Sevier, C.S. A conserved cysteine within the ATPase domain of the endoplasmic reticulum chaperone BiP is necessary for a complete complement of BiP activities. J. Mol. Biol. 2016, 428, 4168–4184.
- Kopp, M.C.; Larburo, N.; Duraiaj, V.; Adams, C.J.; Ali, M.M.U. UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat. Stuct. Mol. Biol. 2020, 26, 1053–1062.
- Haßdenteufel, S.; Johnson, N.; Paton, A.W.; Paton, J.C.; High, S.; Zimmermann, R. Chaperone-mediated Sec61 channel gating during ER import of small precursor proteins overcomes Sec61 inhibitor-reinforced energy barrier. Cell Rep. 2018, 23, 1373–1386.
- Hamman, B.D.; Hendershot, L.M.; Johnson, A.E. BiP maintains the permeability barrier of the ER membrane by sealing the lumenal end of the translocon pore before and early in translocation. Cell 1998, 92, 747–758.
- Alder, N.A.; Shen, Y.; Brodsky, J.L.; Hendershot, L.M.; Johnson, A.E. The molecular mechanism underlying BiP-mediated gating of the Sec61 translocon of the endoplasmic reticulum. J. Cell Biol. 2005, 168, 389–399.
- Hennessy, F.; Nicoll, W.S.; Zimmermann, R.; Cheetham, M.E.; Blatch, G.L. Not all J domains are created equal: Implications for the specificity of Hsp40-Hsp70 interactions. Protein Sci. 2005, 14, 1697–1709.
- Chung, K.T.; Shen, Y.; Hendershot, L.M. BAP, a mammalian BiP associated protein, is a nucleotide exchange factor that regulates the ATPase activity of BiP. J. Biol. Chem. 2002, 277, 47557–47563.
- Lin, H.-Y.; Masso-Welch, P.; Di, Y.-P.; Cai, J.-W.; Shen, J.-W.; Subjeck, J.R. The 170-kDa glucose-regulated stress protein is an endoplasmic reticulum protein that binds immunoglobulin. Mol. Biol. Cell 1993, 4, 1109–1119.
- Kitao, Y.; Hashimoto, K.; Matsuyama, T.; Iso, H.; Tamatani, T.; Hori, O.; Stern, D.M.; Kano, M.; Ozawa, K.; Ogawa, S. ORP150/HSP12A regulates purkinje cell survival: A role for endoplasmic reticulum stress in cerebellar development. J. Neurosci. 2004, 24, 1486–1496.
- Weitzmann, A.; Volkmer, J.; Zimmermann, R. The nucleotide exchange factor activity of Grp170 may explain the non-lethal phenotype of loss of Sil1 function in man and mouse. FEBS Lett. 2006, 580, 5237–5240.
- Behnke, J.; Feige, M.J.; Hendershot, L.M. BiP and its nucleotide exchange factors Grp170 and Sil1: Mechanisms of action and biological functions. J. Mol. Biol. 2015, 427, 1589–1608.
- Shomura, Y.; Dragovic, Z.; Chang, H.C.; Tzvetkov, N.; Young, J.C.; Brodsky, J.L.; Guerriero, V.; Hartl, F.U.; Bracher, A. Regulation of Hsp70 function by HspBP1: Structural analysis reveals an alternate mechanism for Hsp70 nucleotide exchange. Mol. Cell 2005, 17, 367–379.
- Polier, S.; Dragovic, Z.; Hartl, F.U.; Bracher, A. Structural basis for the cooperation of Hsp70 and Hsp110 chaperones in protein folding. Cell 2008, 133, 1068–1079.
- Haigh, N.G.; Johnson, A.E. A new role for BiP: Closing the aqueous translocon pore during protein integration into the ER membrane. J. Cell Biol. 2002, 156, 261–270.
- Madeo, F.; Kroemer, G. Intricate links between ER stress and apoptosis. Mol. Cell 2009, 33, 669–670.
- Malhotra, J.D.; Kaufman, R.J. ER stress and its functional link to mitochondria: Role in cell survival and death. Cold Spring Harb. Perspect. Biol. 2011, 3, a004424.
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190.
- Ron, D.; Harding, H.P. Protein-folding homeostasis in the endoplasmic reticulum and nutritional regulation. Cold Spring Harb. Perspect. Biol. 2012, 4, a013177.
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 2013, 5, a013169.
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086.
- Vishnu, N.; Jadoon Khan, M.; Karsten, F.; Groschner, L.N.; Waldeck-Weiermair, M.; Rost, R.; Hallström, S.; Imamura, H.; Graier, W.F.; Malli, R. ATP increases within the lumen of the endoplasmic reticulum upon intracellular Ca2+ release. Mol. Biol. Cell 2014, 25, 368–379.
- Klein, M.-C.; Zimmermann, K.; Schorr, S.; Landini, M.; Klemens, P.; Altensell, J.; Jung, M.; Krause, E.; Nguyen, D.; Helms, V.; et al. AXER is an ATP/ADP exchanger in the membrane of the endoplasmic reticulum. Nat. Commun. 2018, 9, 3489.
- Yong, J.; Bischof, H.; Burgstaller, S.; Siirin, M.; Murphy, A.; Malli, R.; Kaufman, R.J. Mitochondria supply ATP to the ER through a mechanism antagonized by cytosolic Ca2+. ELife 2019, 8, e49682.
- Zimmermann, R.; Lang, S. A little AXER ABC: ATP, BiP, and Calcium form a triumvirate orchestrating energy homeostasis of the endoplasmic reticulum. Contact 2020.
- Amin-Wetzel, N.; Saunders, R.A.; Kamphuis, M.J.; Rato, C.; Preissler, S.; Harding, H.P.; Ron, D. A J-protein co-chaperone recruits BiP to monomerize IRE1 and repress the unfolded protein response. Cell 2017, 171, 1625–1637.
- Haßdenteufel, S.; Klein, M.-C.; Melnyk, A.; Zimmermann, R. Protein transport into the human ER and related diseases: Sec61-channelopathies. Biochem. Cell Biol. 2014, 92, 499–509.
- Luesch, H.; Paavalainen, V.O. Natural products as modulators of eukayotic secretion. Nat. Prod. Rep. 2020, 37, 717.
- Zehner, M.; Marschall, A.L.; Bos, E.; Schloetel, J.-G.; Kreer, C.; Fehrenschild, D.; Limmer, A.; Ossendorp, F.; Lang, T.; Koster, A.J.; et al. The translocon protein Sec61 mediates antigen transport from endosomes in the cytosol for cross-presentation to CD8+ T cells. Immunity 2015, 42, 850–863.