Prostate-specific membrane antigen (PSMA) is a transmembrane protein that is overexpressed in prostate cancer and correlates with the aggressiveness of the disease. PSMA is a promising target for positron emission tomography (PET) imaging and theranostics in prostate cancer patients validated in recent prospective trials. Several clinical trials are currently ongoing to define the role of PSMA targeting radioligands in different settings and to evaluate the potential of other PSMA-based therapeutic modalities in prostate cancer.
- castration resistant prostate cancer (CRPC)
- molecular imaging
- positron emission tomography (PET)
- prostate-specific membrane antigen (PSMA)
1. Introduction
Prostate cancer is the second most commonly diagnosed cancer in men and the sixth leading cause of cancer-related deaths among men worldwide although incidence and mortality of prostate cancer vary depending on the country [1]. Incidence and mortality rates have been on the decline or have stabilized recently particularly in high-income countries. Although the prognosis for localized prostate cancer is good, with 5-year survival rates above 90%, recurrence can occur after radical therapy, and many patients have metastases at the time of primary diagnosis [2]. Conventional 99mTc-bone scintigraphy and computed tomography (CT) showed limited diagnostic accuracy to detect and localize disease or treatment response in many cases. In particular, they are insensitive in the event of biochemical relapse with a rising prostate-specific antigen (PSA) after radical treatment, presenting an unmet need for better methods.
The first-line treatment option for metastatic prostate cancer is androgen deprivation therapy (ADT) that can be combined with docetaxel chemotherapeutic agent or drugs interfering with androgen signaling in early castration naïve state, but eventually, the lethal castration-resistant disease develops [2,3][2][3]. The therapeutic landscape of metastatic prostate cancer has evolved during recent years to extend survival. Prostate-specific membrane antigen (PSMA) is a promising clinically validated target for expression-based imaging and therapies, but a deeper understanding of the underlying biology is needed to optimize its use. Evidence from the first randomized phase III trial supports the benefit of therapeutically targeting PSMA [4].
2. The Role of PSMA in the Biology of Prostate Cancer
2.1. Gene and Protein Structure of PSMA
PSMA, also known as glutamate carboxypeptidase II (GCPII), N-acetylaspartylglutamate peptidase, and N-acetyl-L-aspartyl-L-glutamate peptidase I (NAALADase I), is a type II transmembrane glycoprotein encoded by FOLH1 (folate hydrolase 1) gene mapped to chromosome 11 short arm (11p11–11p12) and contains 19 exons, encoding a protein of 750 amino acids and a molecular weight of approximately 100 kDa [5,6][5][6].
PSMA contains a catalytic domain responsible for both NAALADase and folate hydrolase activity and belongs to the M28 metalloprotease family that contains aminopeptidases and carboxypeptidases. PSMA protein has a large extracellular domain, a short transmembrane domain, and a short cytoplasmic tail [6,7,8][6][7][8]. The extracellular domain consists of three subdomains: the protease, the apical domain, and the dimerization domain, which are all necessary for substrate binding [9,10][9][10] (Figure 1A). In the binding cavity of PSMA, the pharmacophore pocket stabilizes glutamate-like moieties using polar and van der Waals interactions [11]. The active site of PSMA contains two catalytic zinc ions coordinated by His377, His553, Asp387, Asp453, and Glu425 [10,12][10][12]. The short intracellular domain contains an internalization motif and interacts with proteins, such as caveolin-1, clathrin, and clathrin adaptor protein 2, enabling PSMA endocytosis via caveolae-dependent mechanisms and via clathrin-coated pits [13,14,15][13][14][15]. Additionally, an interaction between actin-binding protein Filamin A (FLNa) and the cytoplasmic tail of PSMA has been shown to decrease the internalization and the enzymatic NAALADase activity of PSMA in vitro [16].
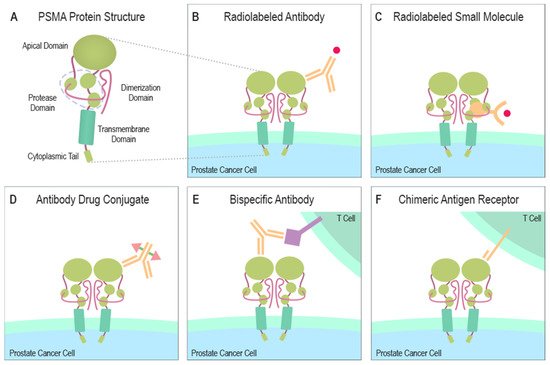
Figure 1. Structure of PSMA and PSMA-targeting therapeutical modalities. (A) The large extracellular portion of PSMA contains the protease domain that cleaves glutamate from NAAG and polyglutamated folates. The cytoplasmic tail interacts with several proteins some of which can induce the endocytosis of PSMA. (B) A radionuclide capable of emitting ionizing radiation due to radioactive decay can be combined with a PSMA-specific antibody to create a PSMA-targeting cytotoxic molecule. (C) A similar function is gained when a small molecule, naturally bound by PSMA, is linked to a radionuclide emitting ionizing radiation. (D) ADCs are altered antibodies carrying therapeutic agents to the targeted protein. Current ADC trials are testing PSMA-targeting antibodies carrying microtubule-disrupting agents (Table 1). (E) Bispecific antibodies can be designed to target PSMA and simultaneously attach to CD3 or CD28 expressed by T cells. (F) Autologous or allogeneic T cells can be engineered to express PSMA-targeting CARs. Current clinical trials are also studying similarly engineered NK cells. CAR T or NK cells can be designed to ignore immunosuppressive signals from the tumor microenvironment by making them insensitive to certain molecules e.g., PD-1. Abbreviations: PSMA = prostate-specific membrane antigen, NAAG = N-acetylaspartylglutamate, ADC = antibody-drug conjugate, CD3 = cluster of differentiation 3, CD28 = cluster of differentiation 28, CAR = chimeric antigen receptor, NK = natural killer cell, PD-1 = programmed cell death protein 1.
2.2. Expression and Function of PSMA in Normal Tissues
In general, only very low levels of PSMA protein expression have been detected in healthy tissues such as the kidney, intestine, salivary glands, and brain, and it seems that prostatic epithelium is the only tissue to express a significant level of PSMA [17,18][17][18]. Despite more than three decades of extensive research, the exact biological role of the human PSMA protein is not fully understood. Several independent research groups have inactivated the PSMA-encoding gene Folh1 in mice to understand the physiological role of the mouse homolog of human PSMA [19,20,21,22][19][20][21][22]. In mice, PSMA is particularly expressed in the brain and kidney according to the Northern blot analyses [23]. Bacich et al. [19] report that PSMA null mice (intron-exon boundary sequences of exons 1 and 2 deleted and stop codons inserted in exon 1 and 2) have similar N-acetyl-L-aspartyl-L-glutamate (NAAG) levels in the brain as compared to wild-type (WT) mice, suggesting genetic redundancy. The null mice developed normally to adulthood but in comparison to WT showed lower susceptibility to peripheral neuropathies and traumatic brain injury [24]. In accordance, Gao et al. [22] reported PSMA null mice (deletion of exons 3 to 5) with normal breeding performance and no obvious phenotype. Vorlová et al. [25] produced PSMA deficient mice by inactivating (deleted exon 11) Folh1 gene using transcription activator-like effector nuclease (TALEN)-mediated mutagenesis. They confirmed that PSMA protein was not expressed and NAAG hydrolyzing activity was lowered, but the PSMA-deficient mice bred and developed normally. They reported that PSMA-deficient aged mice might have an increased propensity for enlarged seminal vesicles compared to WT, but no other obvious phenotype in the urogenital system. In contrast, Tsai et al. [20] reported that PSMA null mice (deletion of exons 9 and 10) died during embryogenesis. The same research group later generated mice also by using the strategy reported by Bacich et al., and again reported that PSMA knockout is embryonically lethal [20,21][20][21]. They suggest that PSMA expression in embryonic stem cells might be important at very early stages of embryonic development, but the reason for the discrepancy with other studies is not clear [21].
In humans, the functional role of PSMA is context-dependent and tissue-specific. PSMA has an enzymatic activity to release glutamate from the substrate as a folate hydrolase and NAALADase [26,27,28][26][27][28]. In the intestine, PSMA detaches glutamates from the C-terminal end of poly-γ-glutamate of the dietary folic-polyglutamates enabling absorption of monoglutamated folates into the enterocytes [29,30][29][30]. In the nervous system, PSMA modulates neuronal signaling by catalyzing the hydrolysis of the neurotransmitter NAAG yielding N-acetyl-aspartate (NAA) and free L-glutamate [31]. Glutamate is the major excitatory neurotransmitter in the nervous system. The released glutamate activates postsynaptic metabotropic (mGluR) and ionotropic (iGluR) receptors of glutamate [32]. In conclusion, although PSMA is a multifunctional protein expressed in some healthy tissues, the most significant expression levels are found in humans in the prostatic epithelium.
2.3. Expression and Function of PSMA in Malignancies
PSMA was first characterized in 1986 by the murine monoclonal antibody 7E11, delivered from mice immunized with partially purified, cell membrane fractions isolated from the human prostate adenocarcinoma cell line LNCaP [7]. Later PSMA has been implicated a role in diseases, such as amyotrophic lateral sclerosis (ALS), schizophrenia, multiple sclerosis, inflammatory bowel disease (IBD), and cancer [33,34,35,36][33][34][35][36]. In cancer, PSMA expression has been detected on the endothelial cells of the neovasculature of several solid tumors, such as renal, bladder, gastric, and colorectal cancer as well as prostate cancer [17,37,38,39][17][37][38][39]. In prostate cancer, PSMA is highly overexpressed at the protein level in cancer cells when compared to normal prostate tissue [40]. Expression level correlates with the aggressiveness of the disease and high PSMA expression levels have been associated with hormone-refractory and metastatic prostate cancer [18,40,41,42,43,44][18][40][41][42][43][44]. However, the expression of PSMA in prostate cancer can be very heterogeneous, and some primary tumors and metastases are negative for PSMA [7,18,40,45][7][18][40][45]. The use of different antibodies in immunohistochemistry with different epitopes can also challenge the interpretation of results [38]. Heterogenous expression of PSMA may be explained by regulation of PSMA by local biological factors and tumor cell microenvironment.
Interestingly, by using a PSMA targeting antibody (YPSMA-1), an inhibitor of the enzymatic activity (2-PMPA) or PSMA null mice as models, it was shown that PSMA regulates endothelial cell invasion into the extracellular matrix without significantly affecting viability, proliferation, or morphogenesis [46]. This suggests that PSMA regulates angiogenesis depending on the enzymatic activity. Mechanistically, laminin-specific integrin β1 activation promotes activation of p21-activated kinase 1 (PAK) to interact with FLNa, disrupting PSMA-FLNa interaction and the enzymatic activity of PSMA. Disruption of PSMA-FLNa interaction causes a reduction in both integrin β1 signaling and PAK activation creating a negative feedback loop. Later, it was recognized that PSMA takes part in a pathway producing pro-angiogenic fragments from extracellular protein laminin, promoting angiogenesis by regulating integrin β1 signaling in endothelial cells [47]. In conclusion, high PSMA expression levels have been detected specifically in prostate cancer cells and in the neovasculature of some solid tumors, making it an attractive target for molecular imaging and therapeutics.
2.4. Functional Role of PSMA in Prostate Cancer
Although PSMA was originally identified in a prostate cancer LNCaP cell line and early on linked to prostate cancer aggressiveness, PSMA is not expressed in many other commonly used commercially available prostate cancer cell lines and preclinical publications have been in part contradictory, demonstrating inhibition of PSMA to either promote or prevent invasion in vitro [48,49][48][49]. First, in 2005, Ghosh et al. [48] showed that transfecting PSMA negative PC3 cells to overexpress wild-type PSMA reduced invasiveness and knocking down endogenous PSMA in LNCaP cells increased invasiveness in the Matrigel invasion assay. Later, they showed using the PSMA-overexpressing transgenic mouse model, that PSMA overexpression increased prostate cancer cell growth in prostate recombinants [49]. Moreover, they repeated the Matrigel invasion assay using low levels of folate and surprisingly observed that ectopic expression of PSMA induced invasiveness in PC3 cells, suggesting that folate levels might modulate the functional consequence to inhibition of PSMA in prostate cancer cells—at least within in vitro. Colombatti et al. also suggested that treating LNCaP cells endogenously expressing PSMA with antibodies against PSMA induced proliferative MAPK pathway activation [50]. The abundance of PSMA in prostate tissue allows increased hydrolysis of the polyglutamated folates yielding glutamate and folate monoglutamate enabling the intake of folates into the cell via proton-coupled folate transporters (PCFT), reduced folate carriers (RFC), or possibly by PSMA itself [51,52][51][52]. Folate is crucial for one-carbon metabolism and is involved in the synthesis of DNA and RNA, and amino acid metabolism [51]. Some studies suggest that decreased folate levels cause epigenetic changes, DNA breaks, translocations, and uracil misincorporation into DNA suggesting a possible carcinogenic role for low folate levels [53,54,55,56][53][54][55][56] but the role of folate in cancer seems complex and also conflicting results exist, suggesting that decreased folate levels are a protective factor against prostate carcinogenesis [57]. Based on a meta-analysis, high blood folate levels associate with increased risk of prostate cancer although the increased dietary and total folate intake do not appear significantly to associate with prostate cancer risk [53].
Folic acid is a synthetic, fully oxidized, monoglutamated version of folate—found for example in vitamin supplements—that can directly be transported into cells while polyglutamated folates have to be hydrolyzed into monoglutamates before being absorbed in the digestive tract. It has been suggested that polyglutamated folate in turn could become available as a substrate for PSMA in particular if intracellular molecules were released by cancer cells undergoing necrotic cell death [58]. Interestingly, tumor necrosis in radical prostatectomies was associated with aggressive features [59]. Treatment of LNCaP cells endogenously expressing PSMA with folic acid induced activation of PI3Kβ-Akt pathway, which was not observed in the presence of inhibitors of either PSMA, PI3Kβ, or mGluRI activity [60]. This suggests that PSMA is capable of activating the PI3K-Akt cell survival pathway in prostate cancer and surprisingly finds mGluRI as the major mediator of folate-induced PI3Kβ-Akt pathway activation by PSMA. Glutamate is a necessary metabolic precursor for other amino acids and nucleotides and is involved in a variety of different transporter and receptor systems that activate proliferative signaling pathways. Glutamate also mobilizes calcium from the endoplasmic reticulum via the activation of mGluR [60]. Expression of glutamate transporters and many of the different iGluRs and mGluRs have been reported in LNCaP and PC3 prostate cancer cell lines [61]. Glutamate deprivation or blockade with mGluR1-antagonist results in significantly decreased cell proliferation, migration, and invasion of prostate cancer cells, leading to apoptotic cell death, demonstrating a potentially important role of glutamate pathway for prostate cancer growth [62]. In prostate cancer patients, serum glutamate levels directly correlate with Gleason Score and aggressiveness [62]. Moreover, high mGluR1 levels in primary and metastatic prostate cancer tissue when compared to benign prostate tissue samples have been detected by immunohistochemistry [62].
Caromile et al. [63] crossed PSMA null mice with a TRAMP transgenic mouse model to investigate in vivo effects of PSMA in prostate carcinogenesis. Prostate cancer progression in a TRAMP transgenic mouse model was less aggressive in PSMA deficient background, suggesting a direct role for PSMA in prostate carcinogenesis. PSMA positive tumors were bigger and had a higher microvessel density when compared to tumors in the PSMA knockout animals. Mechanistically, PSMA was shown to interact intracellularly with a scaffolding protein receptor for activated C kinase 1 (RACK1) and disrupt the interaction between insulin-like growth factor 1 receptor (IGF1-R) and integrin β1, resulting in a switchlike change from activation of the proliferation-associated MAPK pathway to activation of a cell survival-associated PI3K-AKT pathway. This suggested one mechanism of how PSMA overexpression could drive prostate cancer growth and indicated that disrupting the interaction between PSMA and the scaffold might have therapeutic potential for patients with PSMA-positive prostate cancer. Mutations targeting phosphatase and tensin homolog (PTEN) or other components of the PI3K-AKT pathway are often found in prostate cancer and crosstalk between the PI3K-AKT pathway and AR signaling has been described [64,65][64][65]. This reciprocal feedback regulation may be involved in resistance to therapeutics resulting in AR or PI3K-AKT inhibition, resulting in the activation of the other pathway when the other one is inhibited [64,65][64][65]. The integrin β1-mediated mechanism linking PSMA to the PI3K-AKT pathway may thus result in a similar reciprocal feedback regulation towards AR signaling. Interestingly, RACK1 has also been shown to interact with AR and inhibit its function, putatively also connecting PSMA to AR signaling [66].
In conclusion, recent studies using different model systems including transgenic mouse models with PSMA null background have elucidated several distinct mechanisms associating PSMA with survival-related signaling pathways with connections to the androgen receptor (AR) signaling in prostate cancer (Figure 2). The newly discovered mechanisms provide new context also for the earlier studies and cooperatively suggest a role for PSMA as an important participant in multiple stages of prostate cancer progression.
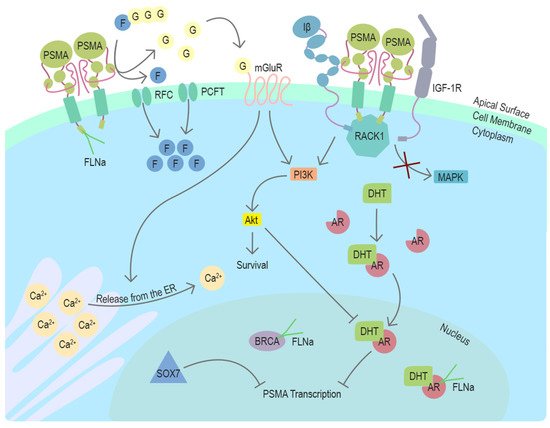
Figure 2. Function and regulators of PSMA in prostate cancer cells. PSMA is a transmembrane protein expressed on the surface of prostate cancer cells. In the dimeric form, PSMA can enzymatically hydrolyze glutamated folates producing glutamate and folates that can enter the cell through RFC or PCFT. Activation of mGluR by glutamate induces the release of calcium from the ER and activates cell survival associated with the PI3K-Akt pathway. In the absence of PSMA, Integrin β (Iβ) and IGF1-1R interact intracellularly with a scaffold protein RACK1 leading to the activation of the MAPK pathway. This interaction is disrupted by PSMA, changing the pathway activation from MAPK to PI3K-Akt in a switchlike manner. DHT is the physiological activator of AR. PSMA transcription is suppressed by AR after activation by DHT. Another potential repressor of PSMA transcription in prostate cancer cells is SOX7, which has been shown to bind to PSME. Cytoskeleton-related protein FLNa interacts with the intracellular domain of PSMA. FLNa interestingly also interacts with AR and BRCA1, and BRCA2. Abbreviations: PSMA = prostate-specific membrane antigen, FLNa = filamin A, G = glutamate, F = folate, RFC = reduced folate carrier, PCFT = proton-coupled folate transporter, mGluR = metabotropic glutamate receptor, ER = endoplasmic reticulum, Iβ = integrin β, IGF-1R = insulin-like growth factor 1 receptor, RACK1 = receptor of activated protein C kinase 1, MAPK = mitogen-activated protein kinase, PI3K = phosphoinositide 3-kinase, Akt/PKB = protein kinase B, DHT = dihydrotestosterone, AR = androgen receptor, PSME = PSMA enhancer, BRCA = breast cancer type 1 or 2 susceptibility protein, SOX7 = SRY-Box transcription factor 7, Ca2+ = calcium ion.
References
- Culp, M.B.; Soerjomataram, I.; Efstathiou, J.A.; Bray, F.; Jemal, A. Recent Global Patterns in Prostate Cancer Incidence and Mortality Rates. Eur. Urol. 2020, 77, 38–52.
- Parker, C.; Castro, E.; Fizazi, K.; Heidenreich, A.; Ost, P.; Procopio, G.; Tombal, B.; Gillessen, S. Prostate cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 1119–1134.
- Sartor, O.; de Bono, J.S. Metastatic Prostate Cancer. N. Engl. J. Med. 2018, 378, 1653–1654.
- Novartis Announces Positive Result of Phase III Study with Radioligand Therapy 177Lu-PSMA-617 in Patients with Advanced Prostate Cancer. Available online: (accessed on 24 March 2021).
- O’Keefe, D.S.; Su, S.L.; Bacich, D.J.; Horiguchi, Y.; Luo, Y.; Powell, C.T.; Zandvliet, D.; Russell, P.J.; Molloy, P.L.; Nowak, N.J.; et al. Mapping, genomic organization and promoter analysis of the human prostate-specific membrane antigen gene. Biochim. Biophy. Acta 1998, 1443, 113–127.
- Israeli, R.S.; Powell, C.T.; Fair, W.R.; Heston, W.D.W. Molecular Cloning of a Complementary DNA Encoding a Prostate-specific Membrane Antigen. Cancer Res. 1993, 53, 227–230.
- Horoszewicz, J.S.; Kawinski, E.; Murphy, G.P. Monoclonal Antibodies to a New Antigenic Marker in Epithelial Prostatic Cells and Serum of Prostatic Cancer Patients. Anticancer. Res. 1987, 7, 927–935.
- Rawlings, N.D.; Barrett, A.J. Structure of Membrane Glutamate Carboxypeptidase. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1997, 1339, 247–252.
- Mindy, I.D.; Melanie, J.B.; Leonard, M.T.; Pamela, J. Bjorkman Crystal Structure of Prostate-Specific Membrane Antigen, a Tumor Marker and Peptidase. Proc. Natl. Acad. Sci. USA 2005, 102, 5981–5986.
- Mesters, J.R.; Barinka, C.; Li, W.; Tsukamoto, T.; Majer, P.; Slusher, B.S.; Konvalinka, J.; Hilgenfeld, R. Structure of Glutamate Carboxypeptidase II, a Drug Target in Neuronal Damage and Prostate Cancer. EMBO J. 2006, 25, 1375–1384.
- Bařinka, C.; Rovenská, M.; Mlčochová, P.; Hlouchová, K.; Plechanovová, A.; Majer, P.; Tsukamoto, T.; Slusher, B.S.; Konvalinka, J.; Lubkowski, J. Structural Insight into the Pharmacophore Pocket of Human Glutamate Carboxypeptidase II. J. Med. Chem. 2007, 50, 3267–3273.
- Klusák, V.; Bařinka, C.; Plechanovová, A.; Mlčochová, P.; Konvalinka, J.; Rulíšek, L.; Lubkowski, J. Reaction Mechanism of Glutamate Carboxypeptidase II Revealed by Mutagenesis, X-ray Crystallography, and Computational Methods. Biochemistry 2009, 48, 4126–4138.
- Anilkumar, G.; Barwe, S.P.; Christiansen, J.J.; Rajasekaran, S.A.; Kohn, D.B.; Rajasekaran, A.K. Association of Prostate-Specific Membrane Antigen with Caveolin-1 and its Caveolae-Dependent Internalization in Microvascular Endothelial Cells: Implications for Targeting to Tumor Vasculature. Microvasc. Res. 2006, 72, 54–61.
- Rajasekaran, S.A.; Anilkumar, G.; Oshima, E.; Bowie, J.U.; Liu, H.; Heston, W.; Bander, N.H.; Rajasekaran, A.K. A Novel Cytoplasmic Tail MXXXL Motif Mediates the Internalization of Prostate-specific Membrane Antigen. Molecular Biol. Cell 2003, 14, 4835–4845.
- Goodman, O.B.; Barwe, S.P.; Ritter, B.; McPherson, P.S.; Vasko, A.; Keen, J.H.; Nanus, D.M.; Bander, N.H.; Rajasekaran, A.K. Interaction of Prostate Specific Membrane Antigen with Clathrin and the Adaptor Protein Complex-2. Int. J. Oncol. 2007, 31, 1199–1203.
- Anilkumar, G.; Rajasekaran, S.A.; Wang, S.; Hankinson, O.; Bander, N.H.; Rajasekaran, A.K. Prostate-specific Membrane Antigen Association with Filamin A Modulates Its Internalization and NAALADase Activity. Cancer Res. 2003, 63, 2645–2648.
- Silver, D.A.; Pellicer, I.; Fair, W.R.; Heston, W.D.; Cordon-Cardo, C. Prostate-Specific Membrane Antigen Expression in Normal and Malignant Human Tissues. Clin. Cancer Res. 1997, 3, 81–85.
- Israeli, R.S.; Powell, C.T.; Corr, J.G.; Fair, W.R.; Heston, W.D. Expression of the Prostate-Specific Membrane Antigen. Cancer Res. 1994, 54, 1807–1811.
- Bacich, D.J.; Ramadan, E.; O’Keefe, D.S.; Bukhari, N.; Wegorzewska, I.; Ojeifo, O.; Olszewski, R.; Wrenn, C.C.; Bzdega, T.; Wroblewska, B.; et al. Deletion of the glutamate carboxypeptidase II gene in mice reveals a second enzyme activity that hydrolyzes N-acetylaspartylglutamate. J. Neurochem. 2002, 83, 20–29.
- Tsai, G.; Dunham, K.S.; Drager, U.; Grier, A.; Anderson, C.; Collura, J.; Coyle, J.T. Early embryonic death of glutamate carboxypeptidase II (NAALADase) homozygous mutants. Synapse 2003, 50, 285–292.
- Han, L.; Picker, J.D.; Schaevitz, L.R.; Tsai, G.; Feng, J.; Jiang, Z.; Chu, H.C.; Basu, A.C.; Berger-Sweeney, J.; Coyle, J.T. Phenotypic characterization of mice heterozygous for a null mutation of glutamate carboxypeptidase II. Synapse 2009, 63, 625–635.
- Gao, Y.; Xu, S.; Cui, Z.; Zhang, M.; Lin, Y.; Cai, L.; Wang, Z.; Luo, X.; Zheng, Y.; Wang, Y.; et al. Mice Lacking Glutamate Carboxypeptidase II Develop Normally but are Less Susceptible to Traumatic Brain Injury. J. Neurochem. 2015, 134, 340–353.
- Bacich, D.J.; Pinto, J.T.; Tong, W.P.; Heston, W.D. Cloning, Expression, Genomic Localization, and Enzymatic Activities of the Mouse Homolog of Prostate-Specific Membrane Antigen/NAALADase/Folate Hydrolase. Mamm. Genome 2001, 12, 117–123.
- Bacich, D.J.; Wozniak, K.M.; Lu, X.-M.; O’Keefe, D.S.; Callizot, N.; Heston, W.D.W.; Slusher, B.S. Mice Lacking Glutamate Carboxypeptidase II are Protected from Peripheral Neuropathy and Ischemic Brain Injury. J. Neurochem. 2005, 95, 314–323.
- Vorlová, B.; Sedlák, F.; Kašpárek, P.; Šrámková, K.; Malý, M.; Zámečník, J.; Šácha, P.; Konvalinka, J. A novel PSMA/GCPII-deficient mouse model shows enlarged seminal vesicles upon aging. Prostate 2019, 79, 126–139.
- Bzdega, T.; Turi, T.; Wroblewska, B.; She, D.; Chung, H.S.; Kim, H.; Neale, J.H. Molecular cloning of a peptidase against N-acetylaspartylglutamate from a rat hippocampal cDNA library. J. Neurochem. 1997, 69, 2270–2277.
- Luthi-Carter, R.; Barczak, A.K.; Speno, H.; Coyle, J.T. Molecular characterization of human brain N-acetylated alpha-linked acidic dipeptidase (NAALADase). J. Pharmacol. Exp. Ther. 1998, 286, 1020–1025.
- Pinto, J.T.; Suffoletto, B.P.; Berzin, T.M.; Qiao, C.H.; Lin, S.; Tong, W.P.; May, F.; Mukherjee, B.; Heston, W.D. Prostate-specific membrane antigen: A novel folate hydrolase in human prostatic carcinoma cells. Clin. Cancer Res. 1996, 2, 1445–1451.
- Chandler, C.J.; Wang, T.T.; Halsted, C.H. Pteroylpolyglutamate hydrolase from human jejunal brush borders. Purification and characterization. J. Biol. Chem. 1986, 261, 928–933.
- Halsted, C.H.; Ling, E.; Luthi-Carter, R.; Villanueva, J.A.; Gardner, J.M.; Coyle, J.T. Folylpoly-γ-glutamate carboxypeptidase from pig jejunum. Molecular characterization and relation to glutamate carboxypeptidase II. J. Biol. Chem. 2000, 275, 30746.
- Robinson, M.B.; Blakely, R.D.; Couto, R.; Coyle, J.T. Hydrolysis of the brain dipeptide N-acetyl-L-aspartyl-L-glutamate. Identification and characterization of a novel N-acetylated alpha-linked acidic dipeptidase activity from rat brain. J. Biol. Chem. 1987, 262, 14498–14506.
- Reiner, A.; Levitz, J. Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 2018, 98, 1080–1098.
- Ghadge, G.D.; Slusher, B.S.; Bodner, A.; Canto, M.D.; Wozniak, K.; Thomas, A.G.; Rojas, C.; Tsukamoto, T.; Majer, P.; Miller, R.J.; et al. Glutamate carboxypeptidase II inhibition protects motor neurons from death in familial amyotrophic lateral sclerosis models. Proc. Natl. Acad. Sci. USA 2003, 100, 9554–9559.
- Bařinka, C.; Rojas, C.; Slusher, B.; Pomper, M. Glutamate Carboxypeptidase II in Diagnosis and Treatment of Neurologic Disorders and Prostate Cancer. Curr. Med. Chem. 2012, 19, 856–870.
- Rahn, K.A.; Watkins, C.C.; Alt, J.; Rais, R.; Stathis, M.; Grishkan, I.; Crainiceau, C.M.; Pomper, M.G.; Rojas, C.; Pletnikov, M.V.; et al. Inhibition of glutamate carboxypeptidase II (GCPII) activity as a treatment for cognitive impairment in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2012, 109, 20101–20106.
- Rais, R.; Jiang, W.; Zhai, H.; Wozniak, K.M.; Stathis, M.; Hollinger, K.R.; Thomas, A.G.; Rojas, C.; Vornov, J.J.; Marohn, M.; et al. FOLH1/GCPII is elevated in IBD patients, and its inhibition ameliorates murine IBD abnormalities. JCI Insight 2016, 1.
- Chang, S.S. Overview of Prostate-Specific Membrane Antigen. Rev. Urol. 2004, 6, S13–S18.
- Chang, S.S.; Reuter, V.E.; Heston, W.D.; Bander, N.H.; Grauer, L.S.; Gaudin, P.B. Five different anti-prostate-specific membrane antigen (PSMA) antibodies confirm PSMA expression in tumor-associated neovasculature. Cancer Res. 1999, 59, 3192–3198.
- Liu, H.; Moy, P.; Kim, S.; Xia, Y.; Rajasekaran, A.; Navarro, V.; Knudsen, B.; Bander, N.H. Monoclonal antibodies to the extracellular domain of prostate-specific membrane antigen also react with tumor vascular endothelium. Cancer Res. 1997, 57, 3629–3634.
- Wright, G.L.; Harley, C. Expression of prostate-specific membrane antigen in normal, benign, and malignant prostate tissues. Urol. Oncol. Semin. Orig. Investig. 1995, 1, 18–28.
- Kawakami, M.; Nakayama, J. Enhanced Expression of Prostate-specific Membrane Antigen Gene in Prostate Cancer as Revealed by in Situ Hybridization. Cancer Res. 1997, 57, 2321–2324.
- Bostwick, D.G.; Pacelli, A.; Blute, M.; Roche, P.; Murphy, G.P. Prostate specific membrane antigen expression in prostatic intraepithelial neoplasia and adenocarcinoma. Cancer 1998, 82, 2256–2261.
- Sweat, S.D.; Pacelli, A.; Murphy, G.P.; Bostwick, D.G. Prostate-specific membrane antigen expression is greatest in prostate adenocarcinoma and lymph node metastases. Urology 1998, 52, 637–640.
- Ross, J.S.; Sheehan, C.E.; Fisher, H.A.G.; Kaufman, R.P.; Kaur, P.; Gray, K.; Webb, I.; Gray, G.S.; Mosher, R.; Kallakury, B.V.S. Correlation of primary tumor prostate-specific membrane antigen expression with disease recurrence in prostate cancer. Clin. Cancer Res. 2003, 9, 6357–6362.
- Mannweiler, S.; Amersdorfer, P.; Trajanoski, S.; Terrett, J.A.; King, D.; Mehes, G. Heterogeneity of prostate-specific membrane antigen (PSMA) expression in prostate carcinoma with distant metastasis. Pathol. Oncol. Res. 2009, 15, 167–172.
- Conway, R.E.; Petrovic, N.; Li, Z.; Heston, W.; Wu, D.; Shapiro, L.H. Prostate-Specific Membrane Antigen Regulates Angiogenesis by Modulating Integrin Signal Transduction. Mol. Cell Biol. 2006, 26, 5310–5324.
- Conway, R.E.; Rojas, C.; Alt, J.; Nováková, Z.; Richardson, S.M.; Rodrick, T.C.; Fuentes, J.L.; Richardson, N.H.; Attalla, J.; Stewart, S.; et al. Prostate-specific membrane antigen (PSMA)-mediated laminin proteolysis generates a pro-angiogenic peptide. Angiogenesis 2016, 19, 487–500.
- Ghosh, A.; Wang, X.; Klein, E.; Heston, W.D.W. Novel role of prostate-specific membrane antigen in suppressing prostate cancer invasiveness. Cancer Res. 2005, 65, 727–731.
- Yao, V.; Parwani, A.; Maier, C.; De Wayne Heston, W.; Bacich, D.J. Moderate Expression of Prostate Specific Membrane Antigen, a Tissue Differentiation Antigen and Folate Hydrolase, Facilitates Prostate Carcinogenesis. Cancer Res. 2008, 68, 9070–9077.
- Colombatti, M.; Grasso, S.; Porzia, A.; Fracasso, G.; Scupoli, M.T.; Cingarlini, S.; Poffe, O.; Naim, H.Y.; Heine, M.; Tridente, G.; et al. The Prostate Specific Membrane Antigen Regulates the Expression of IL-6 and CCL5 in Prostate Tumour Cells by Activating the MAPK Pathways1. PLoS ONE 2009, 4, e4608.
- Tisman, G. Modulation of One-Carbon Metabolism by B Vitamins: Implications for Transformation and Progression of Prostate Cancer. Prostate Cancer Bench Bedside 2011.
- Yao, V.; Berkman, C.E.; Choi, J.K.; O’Keefe, D.S.; Bacich, D.J. Expression of prostate-specific membrane antigen (PSMA), increases cell folate uptake and proliferation and suggests a novel role for PSMA in the uptake of the non-polyglutamated folate, folic acid. Prostate 2010, 70, 305–316.
- Tio, M.; Andrici, J.; Cox, M.R.; Eslick, G.D. Folate intake and the risk of prostate cancer: A systematic review and meta-analysis. Prostate Cancer Prostatic Dis. 2014, 17, 213–219.
- Blount, B.C.; Mack, M.M.; Wehr, C.M.; MacGregor, J.T.; Hiatt, R.A.; Wang, G.; Wickramasinghe, S.N.; Everson, R.B.; Ames, B.N. Folate Deficiency Causes Uracil Misincorporation into Human DNA and Chromosome Breakage: Implications for Cancer and Neuronal Damage. Proc. Natl. Acad. Sci. USA 1997, 94, 3290–3295.
- Bistulfi, G.; Vandette, E.; Matsui, S.; Smiraglia, D.J. Mild folate deficiency induces genetic and epigenetic instability and phenotype changes in prostate cancer cells. BMC Biol. 2010, 8, 6.
- Stempak, J.M.; Sohn, K.; Chiang, E.; Shane, B.; Kim, Y. Cell and stage of transformation-specific effects of folate deficiency on methionine cycle intermediates and DNA methylation in an in vitro model. Carcinogenesis 2005, 26, 981–990.
- Bistulfi, G.; Foster, B.A.; Karasik, E.; Gillard, B.; Miecznikowski, J.; Dhiman, V.K.; Smiraglia, D.J. Dietary folate deficiency blocks prostate cancer progression in the TRAMP model. Cancer Prev. Res. 2011, 4, 1825–1834.
- Yao, V.; Bacich, D.J. Prostate specific membrane antigen (PSMA) expression gives prostate cancer cells a growth advantage in a physiologically relevant folate environment in vitro. Prostate 2006, 66, 867–875.
- Acosta, A.M.; Al Rasheed, M.R.; Rauscher, G.H.; Vormittag, E.; Mon, K.S.; Sharif, A.; Kajdacsy-Balla, A.; Mohapatra, G. Tumor necrosis in radical prostatectomies with high-grade prostate cancer is associated with multiple poor prognostic features and a high prevalence of residual disease. Hum. Pathol. 2018, 75, 1–9.
- Kaittanis, C.; Andreou, C.; Hieronymus, H.; Mao, N.; Foss, C.A.; Eiber, M.; Weirich, G.; Panchal, P.; Gopalan, A.; Zurita, J.; et al. Prostate-specific membrane antigen cleavage of vitamin B9 stimulates oncogenic signaling through metabotropic glutamate receptors. J. Exp. Med. 2018, 215, 159–175.
- Pissimissis, N.; Papageorgiou, E.; Lembessis, P.; Armakolas, A.; Koutsilieris, M. The glutamatergic system expression in human PC-3 and LNCaP prostate cancer cells. Anticancer Res. 2009, 29, 371–377.
- Koochekpour, S.; Majumdar, S.; Azabdaftari, G.; Attwood, K.; Scioneaux, R.; Subramani, D.; Manhardt, C.; Lorusso, G.D.; Willard, S.S.; Thompson, H.; et al. Serum Glutamate Levels Correlate with Gleason Score and Glutamate Blockade Decreases Proliferation, Migration, and Invasion and Induces Apoptosis in Prostate Cancer Cells. Clin. Cancer Res. 2012, 18, 5888–5901.
- Caromile, L.A.; Dortche, K.; Rahman, M.M.; Grant, C.L.; Stoddard, C.; Ferrer, F.A.; Shapiro, L.H. PSMA redirects cell survival signaling from the MAPK to the PI3K-AKT pathways to promote the progression of prostate cancer. Sci. Signal 2017, 10.
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586.
- Mulholland, D.J.; Tran, L.M.; Li, Y.; Cai, H.; Morim, A.; Wang, S.; Plaisier, S.; Garraway, I.P.; Huang, J.; Graeber, T.G.; et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell 2011, 19, 792–804.
- Rigas, A.C.; Ozanne, D.M.; Neal, D.E.; Robson, C.N. The scaffolding protein RACK1 interacts with androgen receptor and promotes cross-talk through a protein kinase C signaling pathway. J. Biol. Chem. 2003, 278, 46087–46093.