The Review covers the synthesis and biological activity of ring-fused benzimidazoles, imidazo[4,5-
The entry covers the synthesis and biological activity of ring-fused benzimidazoles, imidazo[4,5-
f
]benzimidazoles and imidazo[5,4-
f
]benzimidazoles. Compounds are bioreductive antitumor agents when the fused benzene part is a quinone or iminoquinone. Reactions are categorized according to the cyclization reaction, and include oxidative cyclizations using the
t
-amino effect. Other cyclizations are onto the 1- and 2-positions of benzimidazoles using cross-dehydrogenative coupling and homolytic aromatic substitution. Many syntheses are transition metal mediated, but others are metal-free using thermal, electrochemical and photochemical conditions.
- green chemistry
- halogen
- heterocycle
- hydrogen peroxide
- imidazole
- iodine
- nitrosobenzene
- oxone
1. Introduction
1.1. Significance and Biological Activity
Benzimidazole is an important heterocyclic pharmacophore. Therapeutic interest was aroused in the 1950s, when 5,6-dimethylbenzimidazole was discovered as a degradation product of vitamin-B12 (Figure 1) [1,2][1][2]. Benzimidazoles possess a wide range of biological activities [3[3][4][5][6],4,5,6], significantly utilized as APIs in medicines [5], and as pesticides [6]. Moreover among the ring-fused benzimidazoles, pyrimido[1,2-a]benzimidazoles were discovered as effective corticotropin releasing factor-1 (CRF-1) receptor antagonists for potential treatment of mental health disorders [7]. A pyrrolo[1,2-a]benzimidazole was found to be the most effective cyclin-dependent kinase 4/6 (CDK4/6) inhibitor evaluated within a series of highly functionalized five to seven-membered alicyclic ring-fused benzimidazoles [8]. Other ring-fused benzimidazoles, the imidazobenzodiazepines were investigated as poly(ADP-ribose) polymerase (PARP-1) inhibitors, allowing reduction of hyperglycemia with neuroprotective effects in animal models [9].
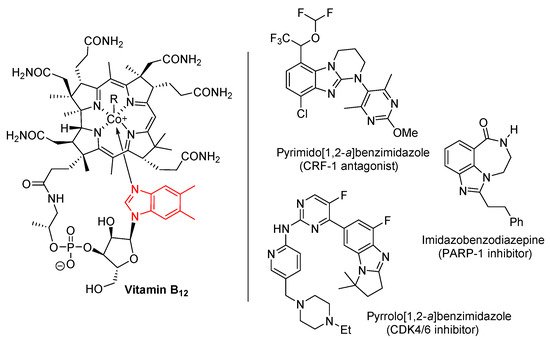
Figure 1.
Benzimidazole in vitamin B
12 and biologically-active ring-fused systems [1,2,3,4,5,6,7,8,9].
and biologically-active ring-fused systems [1][2][3][4][5][6][7][8][9].
The most widely studied of the applications of ring-fused benzimidazoles and imidazobenzimidazoles is as bioreductive antitumor agents, when the fused benzene part is an iminoquinone or quinone functionality (Figure 2, Figure 3, Figure 4 and Figure 5). Indole-based natural products often inspire analogue synthesis, including imidazoquinoxalinone analogues of the marine natural product, wakayin [10], a topoisomerase I inhibitor (Figure 2).
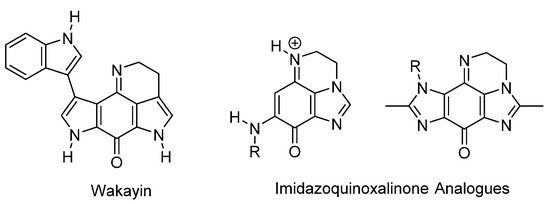
Figure 2. Imidazole-based wakayin analogues [10].
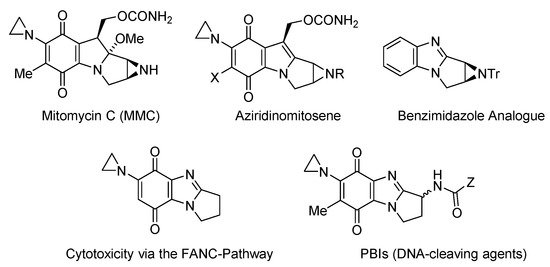
Figure 3. Aziridinyl-functionalized antitumor agents [11,12,13,14,15,16,17,18][11][12][13][14][15][16][17][18].
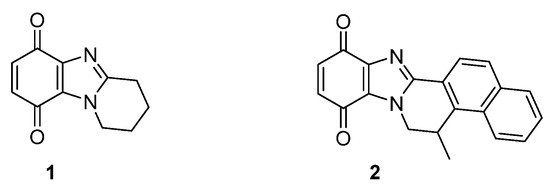
Figure 4. Benzimidazolequinones targeting over-expressed reductases in solid tumors [21,22,23].
Benzimidazolequinones targeting over-expressed reductases in solid tumors [19][20][21].
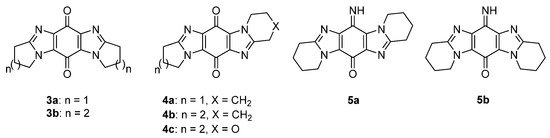
Figure 5. Imidazobenzimidazolequinones and iminoquinones [24,25,26,27,28,29].
Imidazobenzimidazolequinones and iminoquinones [22][23][24][25][26][27].
Mitomycin C (MMC) has, however, attracted most interest, as the archetypal bioreductive antitumor antibiotic, which cross-links to DNA [11]. Incorporation of the DNA-alkylating aziridinyl moiety leads to cytotoxicity via the FANC DNA-repair pathway [12[12][13],13], and the benzimidazole analogue of the bioactivated form of MMC (aziridinomitosene) was prepared and evaluated against breast cancer cell lines [14] (Figure 3). Skibo has published extensively on aziridinylpyrrolo[1,2-a]benzimidazolequinones (PBIs) as DNA cleaving agents with analogues showing melanoma-specific cytotoxicity [15,16,17,18][15][16][17][18].
One or two-electron reductases are responsible for bioreductive activation. NADPH-cytochrome c (P450) reductase is predominant under hypoxic conditions with the one-electron reduction reversed by oxygen [19][28]. Many solid tumors also over-express the obligatory two-electron reductase NAD(P)H:quinone oxidoreductase 1 (NQO1, also known as DT-diaphorase), which is a popular target for anti-cancer studies [20][29]. Many anti-cancer agents do not contain conventional DNA damaging functionality, and cytotoxicity may be due to the formation of reactive oxygen species. Pyrido[1,2-a]benzimidazolequinone 1 (Figure 4) is more than 300 times more cytotoxic under hypoxic conditions than the clinical drug, MMC (Figure 3), with cytotoxicity for alicyclic ring-fused benzimidazoles correlated to reductive potentials [21,22][19][20]. Highly conjugated naphthyl fused benzimidazolequinone 2 leads to increased stability of reduced intermediates leading to specificity towards human cancer cell lines over-expressing NQO1 [23][21].
Ring-fused imidazo[4,5-f]benzimidazolequinones 3a and 3b are NQO1 substrates [24[22][23],25], with 3a, at the National Cancer Institute (NCI), showing specificity towards the killing of melanoma cell lines (Figure 5) [24][22]. Our group was the first to provide viable synthetic protocols for accessing ring-fused imidazo[5,4-f]benzimidazoles, enabling evaluation of quinone and iminoquinone derivatives for toxicity against cancer cell lines [26,27,28,29][24][25][26][27]. Compared to alicyclic ring-fused analogues 4a and 4b, the oxygen atom of the 1,4-oxazino ring was found to increase toxicity of 4c [27][25]. Iminoquinone 5a isolated from the Fremy oxidation to prepare 4b, was unexpectedly the most potent imidazobenzimidazole, with more than 12 times greater cytotoxicity towards a prostate cancer cell line (DU145) than a normal fibroblast cell line (GM00637) [26][24]. More intensive cytotoxicity assays, computational docking, and NCI COMPARE analysis on 5a, revealed good correlation with NQO1 [28][26]. In contrast, isomeric imidazo[4,5-f]benzimidazole 5b was inactive against the NCI 60 cell line panel [29][27].
1.2. Available Synthetic Methods
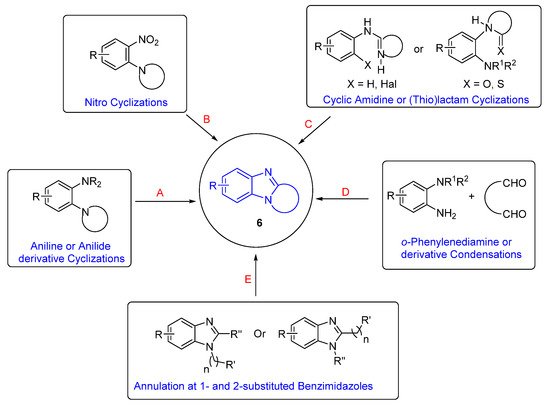
The categories of syntheses of ring-fused benzimidazoles 6 are according to the cyclization reaction (Scheme 1). Oxidative cyclizations from aniline or anilide derivatives is the most studied route (Route A) and is presented in context with the plethora of other syntheses that build the benzimidazole moiety (Routes B–D). The section on Route A is sub-divided into syntheses of benzimidazole and imidazobenzimidazole scaffolds. Lastly, there is a section on syntheses, which begin with the benzimidazole moiety (Route E), sub-divided according to reaction (type) conditions. This is not an exhaustive review, and the reader should consult reviews on polycyclic benzimidazoles for comprehensive lists of syntheses [30,31,32,33][30][31][32][33]. Since the late 1990s, Aldabbagh et al. have worked on the discovery of new ring-fused benzimidazoles and synthetic methods, and the collated articles related to their research are reviewed herein. A reviewer recommended a Scifindern search of “benzimidazole-fused”, which was completed, and significant references are incorporated. For brevity, full papers are cited and not the preceding communication article. A historical perceptive is taken and analysis of the most significant contributions to the field is carried out. In particular, methodology that forms a variety of ring-fused benzimidazoles is of interest, rather than procedures that give mainly the benzimidazole core.
Scheme 1.
Categorizing available synthetic methods according to the cyclization reaction A-E.
2. Syntheses of Ring-Fused Benzimidazoles and Imidazobenzimidazoles
2.1. Oxidations of o-Cycloaminoanilines and Anilide Derivatives (Route A)
There are distinct differences in the reaction mechanisms and conditions for ring-fused benzimidazole and imidazobenzimidazole formation warranting sub-division. Benzimidazoles form by oxidative cyclization of anilines via nitrosobenzene intermediates; in contrast, cyclization to give the ring-fused imidazobenzimidazole must begin from anilides and proceed via amine N-oxide intermediates under acidic conditions.
2.1.1. Forming Ring-Fused Benzimidazoles
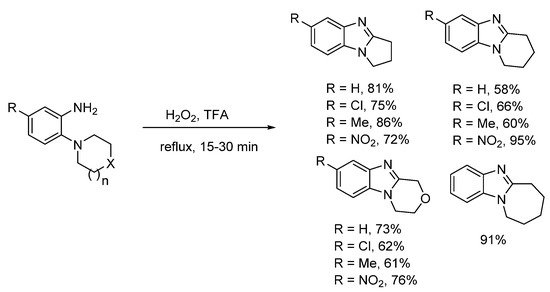
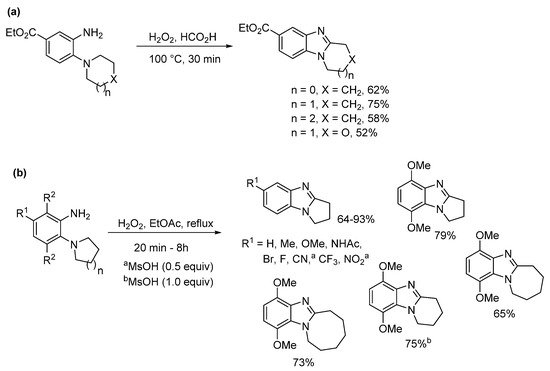
In 1908, Spiegel and Kaufmann reported that Caro’s acid (peroxymonosulfuric acid, H2SO5) oxidized 5-nitro-2-(piperidin-1-yl)aniline to 7-nitro-1,2,3,4-tetrahydropyrido[1,2-a]benzimidazole [34]. In the absence of the nitro-substituent, no oxidative cyclization occurred. Caro’s acid was already known to oxidize anilines to nitrosobenzenes [35], so supporting the idea of a nitroso intermediate. The prominent 20th century chemist and creator of Adam’s catalyst, Roger Adams with Nair refined this methodology, and accessed a range of five to seven-membered ring-fused benzimidazoles in good to high yields using peroxytrifluoroacetic acid generated in situ from H2O2 and trifluoroacetic acid (TFA) (Scheme 2) [36]. Six-membered cyclization yields were higher, when the anilines contained a nitro-substituent. Meth-Cohn and Suschitzky [37] soon refuted the observation made by Nair and Adams that acyl derivatives do not undergo cyclization to give benzimidazoles. These workers showed a range of anilide derivatives (formyl, acetyl and benzoyl) underwent oxidative cyclizations using peroxytrifluoroacetic acid or performic acid (H2O2 and HCO2H). Meth-Cohn preferred the use of o-cyclic amine substituted anilides as substrates for making ring-fused benzimidazoles [37,38][37][38]. Meth-Cohn commented that Nair and Adams [36] had possibly formed the anilide in situ, due to the initial addition of TFA followed by H2O2 [38]. Mechanisms were proposed for benzimidazole formation from anilide, via an amine-N-oxide rather than the nitroso intermediate (see Section 2.1.2). Later the use of anilide derivatives as substrates for preparation of ring-fused benzimidazoles would become commonplace (Scheme 3) [13,15,27,37,38,39,40][13][15][25][37][38][39][40].
Scheme 2. Nair and Adams oxidative cyclizations of anilines [36].
Scheme 3. Acetanilide cyclizations onto (a) azepino- and azocino- [39], and (b) spirocyclic oxetane- [40] derivatives.
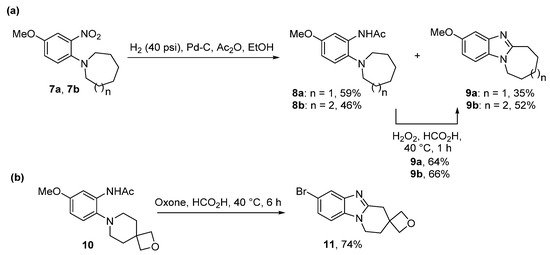
Significant amounts of seven- and eight-membered ring-fused [1,2-a]benzimidazoles 9a and 9b were formed from nitrobenzenes 7a and 7b during the one-pot catalytic hydrogenation and acetylation to give acetamides 8a and 8b (Scheme 3a) [39]. It seemed that the conformation of these large alicyclic rings favored advantageous cyclization of nitroso intermediates formed during the hydrogenation process. Acetamides 8a and 8b were cyclized using performic acid to 9a and 9b in good yields, with the former transformed to 3-aziridinylazepino[1,2-a]benzimidazolequinone [13,39][13][39]. A readily available and safer alternative to Caro’s acid is Oxone (2KHSO5·KHSO4·K2SO4) [41]. Due to the absence of organic waste products, Oxone in the presence of formic acid gives ring-fused benzimidazole [27,40][25][40] and imidazobenzimidazole [27][25] adducts without the requirement for chromatography. 2-Oxa-7-azaspiro[3.5]nonane acetamide 10 gave the spirocyclic oxetane ring-fused benzimidazole 11 in good yield by simple organic extraction from the basified aqueous mixture (Scheme 3b) [40].
Preparations of ring-fused benzimidazoles using o-cyclic amine substituted anilines with performic acid compare favorably with the derivative anilide reaction, with Smalley et al. reporting moderate to good yields for five- to seven-membered adducts (Scheme 4a) [42]. In a more recent study, recyclable ethyl acetate (EtOAc) replaced formic acid, with aqueous effluent, organic-aqueous extraction and chromatography avoided for the preparation of pyrrolo[1,2-a]benzimidazoles from commercial o-(pyrrolidin-1-yl)anilines (Scheme 4b) [43]. Although, the presence of strong electron-withdrawing substituents (NO2, CN) and the six-membered cyclization required methanesulfonic acid (MsOH) to reach high yields. However, MsOH is a green acid undergoing biodegradation by forming CO2 and sulfate [44].
Alternatives to peroxide-based oxidizing systems, include MnO2 in cold chloroform, but yields of ring-fused benzimidazoles from o-cycloaminoanilines were 15–20% due to presumed formation of azo-compounds [45]. Möhrle and Gerloff reported the use of a Hg(II) EDTA complex to deliver ring-fused benzimidazoles, in quantitative yield, apart from the morpholino compound, synthesized in 47% yield (Scheme 5) [46].
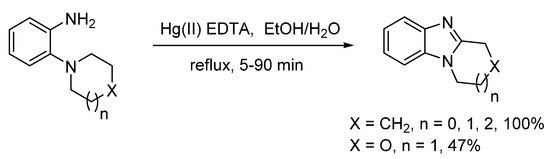
Scheme 5. Hg(II)-mediated oxidative synthesis [46].
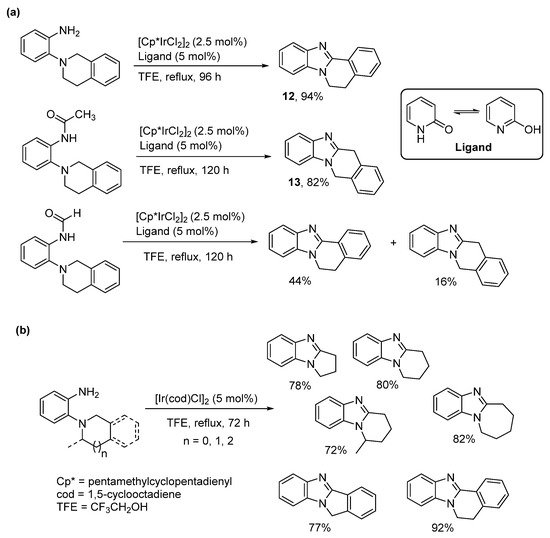
The cross dehydrogenative coupling (CDC) involves forming the C-N bond directly from C-H and N-H bonds under oxidative conditions with a formal loss of H2, in a process often catalyzed by transition metals. CDC is used to describe pentamethylcyclopentadienyl Ir(III)dichloride ([Cp*IrCl2]2) catalyzed oxidative cyclization of o-tetrahydroisoquinoline substituted aniline derivatives (Scheme 6a) [47]. The bulk around the primary amine dictated regioselectivity. The o-cyclic amine substituted aniline gave the benzimidazo[2,1-a]isoquinoline 12, while the more hindered acetamide derivative gave the alternative kinetic product 13. The formamide has less steric bulk than the acetamide forming a mixture of the thermodynamic and kinetic products. The reaction was extended to the synthesis of pyrrolo-, pyrido-, and azepino-[1,2-a]benzimidazoles, without the requirement for a ligand (Scheme 6b), but was less successful for making morpholino- and piperazino-ring-fused analogues [48].
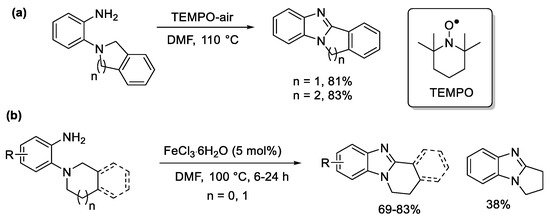
The isoindoline and tetrahydroisoquinoline substrates are the easiest to oxidize at high temperatures, including in the presence of TEMPO in air (Scheme 7a) [49], and catalytic iron(III) [50]. The latter gave the highest yields for the benzimidazo[2,1-a]isoquinoline systems (Scheme 7b).
Aniodic oxidation gave the required iminium ion 14 for cyclization (Scheme 8) [51]. The electrolyte was n-Bu4NPF6 (20 mol%), and the anode is reticulated vitreous carbon (RVC), and Pt is the cathode in an undivided cell at a constant current of 10 mA. A Russian team reported the electrochemical oxidative cyclization with reduction of nitrobenzene for cyclization onto an o-piperidinyl-substituent to give pyrido[1,2-a]benzimidazoles [52].
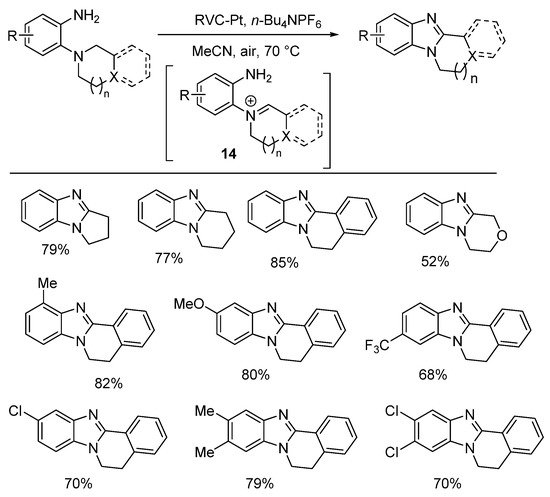
Scheme 8. Electrochemical oxidative cyclizations [51].
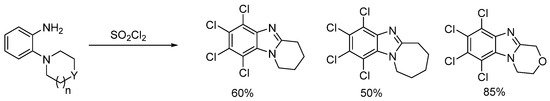
In the early 1970s, o-cyclic amine substituted anilines reacted in neat sulfuryl chloride in an oxidative cyclization with concomitant tetrachlorination of the activated aromatic nucleus (Scheme 9) [53]. The attempt to tetrachlorinate the o-pyrrolo substituted aniline analogue led to an inseparable mixture of mono-, di-, and trichloro ring-fused benzimidazoles.
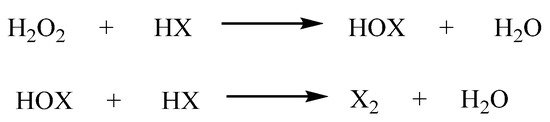
More recently, we heralded the use of H2O2 with hydrohalic acid (HX), as a convenient oxidizing and halogenation system for organic synthesis [54,55,56][54][55][56]. The system allows in situ generation of Cl2 and Br2, with the only by-product, being water (Scheme 10). The salt of hypochlorous acid (HOCl) is the active ingredient in domestic bleach and is the intermediate of the reaction between H2O2 and HCl.
Domestic bleach gave cyclization and dichlorination of aniline 15 in 56% yield (Scheme 11a), with the lower yield attributed to the requirement for chromatography to separate the additives in the bleach [54]. Moreover, using H2O2/HX a library of selectively dichlorinated and dibrominated ring-fused benzimidazoles was prepared in high yields from commercial o-cyclic amine substituted anilines, with most cases not requiring chromatography (Scheme 11b,c). 5-Fluoro-2-piperidinylaniline was an exception, giving significant amounts of cyclization with monochlorination or monobromination. Bromination tended to be slower than chlorination, and tribrominated product 16 was isolated for the o-(pyrrolidin-1-yl)aniline, due to difficulties in cleanly isolating 5,7-dibromopyrrolo[1,2-a]benzimidazole (Scheme 11d).
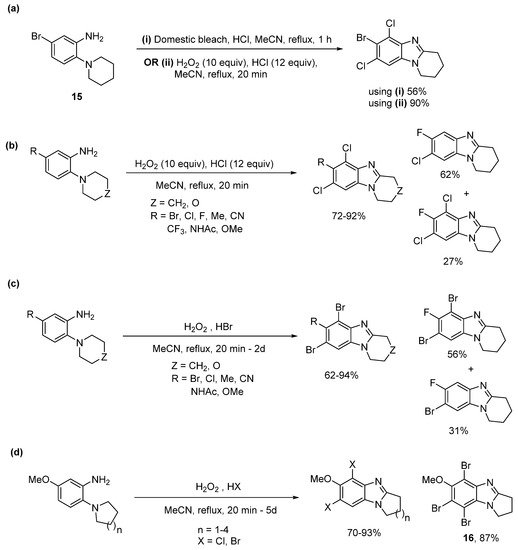
Scheme 11. One-pot ring closure with selective halogenation using (a) domestic bleach or H2O2/HCl, (b) H2O2/HCl, and (c) H2O2/HBr, and (d) five to eight-membered ring-fused adducts [54].
3,6-Dimethoxy-2-(cycloamino)anilines underwent 6-electron oxidations to afford a variety of ring-fused halogenated benzimidazolequinones, when using higher amounts of HCl or HBr relative to H2O2 (Scheme 12a) [55]. The active species is the elemental halogen (X2) with water required for quinone formation (Scheme 12b). When less in situ halogen was generated, using [H2O2] > [HX], the 4-electron oxidation occurred, to give ring-fused halogenated benzimidazoles (Scheme 12c).
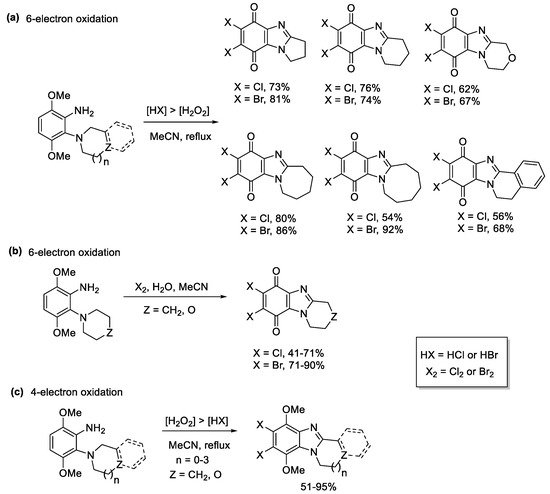
Scheme 12. One-pot transformations of anilines to halogenated ring-fused benzimidazolequinones using (a) H2O2/HX, (b) Cl2 and Br2 with water, and (c) H2O2/HX without quinone formation [55].
The use of hydroiodic acid (HI) is preferred when oxidative cyclization is required without halogenation [56], due to the relatively smaller electrophilicity of iodine [57]. Five- and seven-membered cyclizations of 3,6-dimethoxy-2-(cycloamino)anilines with H2O2 and a catalytic amount of HI in EtOAc proceeded in high yield (Scheme 13a), but 1,4,6,9-tetramethoxyphenazine 17, was unexpectedly formed, as an orange precipitate from six-membered cyclizations (Scheme 13a,b) [56]. The absence of phenazine 17 from the five- and seven-membered cyclizations was consistent with previous observations that six-membered oxidative cyclizations are more difficult [36,43][36][43]. The formation of 17 was optimized by reducing the amount of EtOAc (the reaction solvent) by four-fold and by decreasing the reaction temperature to room temperature (Scheme 14). Moreover, the isolation of 17 was indicative of a nitrosobenzene intermediate in the conversion of o-(cycloamino)anilines to ring-fused benzimidazoles via the so-called t-amino effect [58]. Syntheses of phenazines involve nitroso intermediates [56,59,60][56][59][60]. Recent evidence in the synthesis of ring-fused benzimidazoles, included GC-MS of the reaction mixture (Scheme 14), after 1 h, which revealed EI-MS fragmentation pattern consistent with intermediate 18 [56].
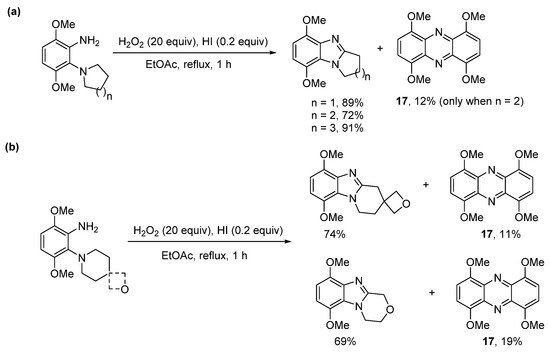
Scheme 13. H2O2/HI-mediated (a) five- to seven-membered cyclizations and (b) six-membered cyclizations with phenazine formation [56].
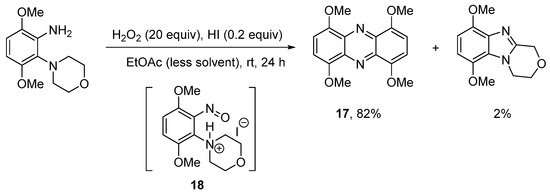
Scheme 14. Optimizing the synthesis of 1,4,6,9-tetramethoxyphenazine 17 [56].
3,4-Dihydro-1H-[1,4]oxazino[4,3-a]benzimidazole can however be prepared in good yield in the absence of acid from 2-(morpholin-4-yl)aniline using Oxone at room temperature (Scheme 15) [61]. Sampling the reaction at short reaction times (within 2 min), gave 4-(2-nitrophenyl)morpholine (20), which formed through advantageous air oxidation of the nitroso intermediate 19. The proposed mechanism postulates that KHSO5 (the active ingredient of Oxone) is catalytic, consistent with the use of catalytic amounts of HI in the H2O2/HI-mediated oxidative cyclization of 3,6-dimethoxy-2-(cycloamino)anilines (Scheme 13 and Scheme 14) [56].
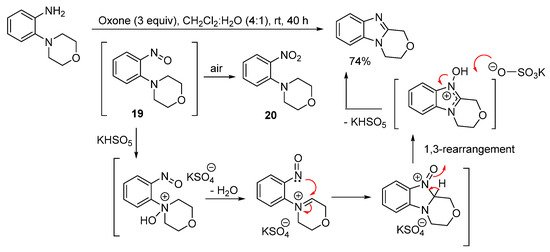
Scheme 15. Oxone-mediated ring-closure of 2-(morpholin-4-yl)aniline [61].
Jana et al. reported one-pot sequential amination of nitrosoarenes with alicyclic amines, followed by oxidative cyclization to give ring-fused benzimidazoles (Scheme 16a–c) [62]. Oxone oxidation of commercial anilines gave the nitrosoarene substrates. Nucleophilic aromatic hydrogen substitution (SNArH) led to the pyrrolo[1,2-a]benzimidazoles, however piperido- and azepino-analogues required nucleophilic aromatic substitution (SNAr) of 2-fluoronitrosobenzene to achieve reasonable yields for the [1,2-a] ring-fused benzimidazoles (Scheme 16a). Nucleophilic ipso-substitution of nitrosonaphthols with cyclic amines and subsequent oxidative cyclization delivered a diverse range of ring-fused naphthoimidazoles (Scheme 16b). The authors proposed a mechanism with mass spectrometry detecting the o-cyclic amine substituted phenyl hydroxylamine 21, which oxidized to the o-cyclic amine substituted nitrosoarene 22 (Scheme 16c). A formal 1,5-hydride shift gave the key iminium ion intermediate 23 for oxidative cyclization [58]. In contrast to our recent work [43[43][56][61],56,61], the nitrosoarene cyclizations, were carried out using elevated temperatures and under acidic conditions [62].
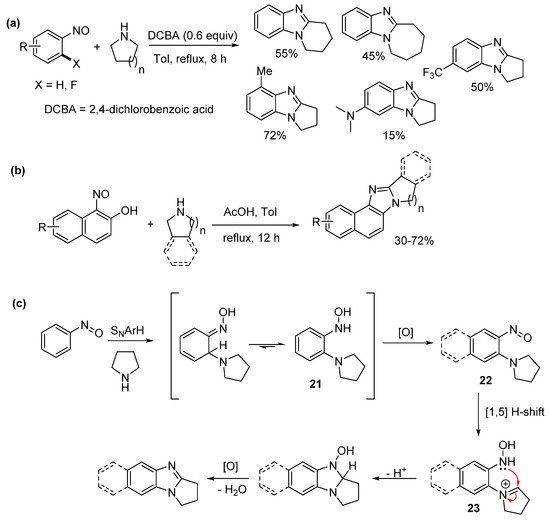
Scheme 16. Sequential aminations of nitrosoarenes to prepare (a) alicyclic ring-fused benzimidazoles, (b) naphthoimidazoles, and (c) the proposed mechanism [62].
References
- Brink, N.G.; Folkers, K. Vitamin B12 VI. 5,6-Dimethylbenzimidazole, a. degradation product of vitamin B12. J. Am. Chem. Soc. 1949, 71, 2951.
- Floyd, J.C.; Holliday, E.R.; Petrow, V. Letters to the Editor. J. Pharm. Pharmacol. 1949, 1, 734–735.
- Yadav, G.; Ganguly, S. Structure activity relationship (SAR) study of benzimidazole scaffold for different biological activities: A mini-review. Eur. J. Med. Chem. 2015, 97, 419–443.
- Keri, R.S.; Hiremathad, A.; Budagumpi, S.; Nagaraja, B.M. Comprehensive review in current developments of benzimidazole-based medicinal chemistry. Chem. Biol. Drug Des. 2015, 86, 19–65.
- Bansal, Y.; Silakari, O. The therapeutic journey of benzimidazoles: A review. Bioorg. Med. Chem. 2012, 97, 419–443.
- Leadbeater, A.J. Plant health management: Fungicides and antibiotics. In Encyclopedia of Agriculture and Food Systems; Van Alfen, N.K., Ed.; Academic Press: Cambridge, MA, USA, 2014; pp. 408–424.
- Kojima, T.; Mochizuki, M.; Takai, T.; Hoashi, Y.; Morimoto, S.; Seto, M.; Nakamura, M.; Kobayashi, K.; Sako, Y.; Tanaka, M.; et al. Discovery of 1,2,3,4-tetrahydropyrimido[1,2-a]benzimidazoles as novel class of corticotropin releasing factor 1 receptor antagonists. Bioorg. Med. Chem. 2018, 26, 2229–2250.
- Wang, Y.; Liu, W.-J.; Yin, L.; Li, H.; Chen, Z.-H.; Zhu, D.-X.; Song, X.-Q.; Cheng, Z.-Z.; Song, P.; Wang, Z.; et al. Design and synthesis of 4-(2,3-dihydro-1H-benzo[d]pyrrolo[1,2-a]imidazol-7-yl)-N-(5-(piperazin-1-ylmethyl)pyridine-2-yl)pyrimidin-2-amine as a highly potent and selective cyclin-dependent kinases 4 and 6 inhibitors and the discovery of structure-activity relationships. Bioorg. Med. Chem. Lett. 2018, 28, 974–978.
- Ferraris, D.; Ficco, R.P.; Dain, D.; Ginski, M.; Lautar, S.; Lee-Wisdom, K.; Liang, S.; Lin, Q.; Lu, M.X.-C.; Morgan, L.; et al. Design and synthesis of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. Part 4: Biological evaluation of imidazobenzodiazepines as potent PARP-1 inhibitors for treatment of ischemic injuries. Bioorg. Med. Chem. 2003, 11, 3695–3707.
- Hoang, H.; Huang, X.; Skibo, E.B. Synthesis and in vitro evaluation of imidazole-based wakayin analogues. Org. Biomol. Chem. 2008, 6, 3059–3064.
- Bass, P.D.; Gubler, D.A.; Judd, T.C.; Williams, R.M. Mitomycinoid alkaloids: Mechanism of action, biosynthesis, total syntheses, and synthetic approaches. Chem. Rev. 2013, 113, 6816–6863.
- O’Donovan, L.; Carty, M.P.; Aldabbagh, F. First synthesis of N-[(aziridin-2-yl)methyl]benzimidazolequinone and analysis of toxicity towards normal and Fanconi anemia cells. Chem. Commun. 2008, 43, 5592–5594.
- Fahey, K.; O’Donovan, L.; Carr, M.; Carty, M.P.; Aldabbagh, F. The influence of the aziridinyl substituent of benzimidazoles and benzimidazolequinones on toxicity towards normal and Fanconi anaemia cells. Eur. J. Med. Chem. 2010, 45, 1873–1879.
- Bonham, S.; O’Donovan, L.; Carty, M.P.; Aldabbagh, F. First synthesis of an aziridinyl fused pyrrolo[1,2-a]benzimidazole and toxicity evaluation towards normal and breast cancer cell lines. Org. Biomol. Chem. 2011, 9, 6700–6706.
- Islam, I.; Skibo, E.B.; Dorr, R.T.; Alberta, D.S. Structure-activity studies of antitumor agents based on pyrrolo[l,2-a]benzimidazoles: New reductive alkylating DNA cleaving agents. J. Med. Chem. 1991, 34, 2954–2961.
- Skibo, E.B.; Gordon, S.; Bess, L.; Boruah, R.; Heileman, M.J. Studies of pyrrolo[1,2-a]benzimidazolequinone DT-diaphorase substrate activity, topoisomerase II inhibition activity, and DNA reductive alkylation. J. Med. Chem. 1997, 40, 1327–1339.
- Craigo, W.A.; LeSueur, B.W.; Skibo, E.B. Design of highly active analogues of the pyrrolo[1,2-a]benzimidazole antitumor agents. J. Med. Chem. 1999, 42, 3324–3333.
- Skibo, E.B.; Jamil, A.; Austin, B.; Hansen, D.; Ghodousi, A. Triple molecular target approach to selective melanoma cytotoxicity. Org. Biomol. Chem. 2010, 8, 1577–1587.
- Lynch, M.; Hehir, S.; Kavanagh, P.; Leech, D.; O’Shaughnessy, J.; Carty, M.P.; Aldabbagh, F. Synthesis by radical cyclization and cytotoxicity of highly potent bioreductive alicyclic ring fused [1,2-a] benzimidazolequinones. Chem. Eur. J. 2007, 13, 3218–3226.
- Hehir, S.; O’Donovan, L.; Carty, M.P.; Aldabbagh, F. Synthesis of dimethyl substituted benzimidazoles containing cyclopropane fused onto five to eight membered [1,2-a] alicyclic rings and influence of methyl group substituents on cytotoxicity of benzimidazolequinones. Tetrahedron 2008, 64, 4196–4203.
- Moriarty, E.; Carr, M.; Bonham, S.; Carty, M.P.; Aldabbagh, F. Synthesis and toxicity towards normal and cancer cell lines of benzimidazolequinones containing fused aromatic rings and 2-aromatic ring substituents. Eur. J. Med. Chem. 2010, 45, 3762–3769.
- Schulz, W.G.; Skibo, E.B. Inhibitors of topoisomerase II based on the benzodiimidazole and dipyrroloimidazobenzimidazole ring systems: Controlling DT-diaphorase reductive inactivation with steric bulk. J. Med. Chem. 2000, 43, 629–638.
- Suleman, A.; Skibo, E.B. A comprehensive study of the active site residues of DT-diaphorase: Rational design of benzimidazolediones as DT-diaphorase substrates. J. Med. Chem. 2002, 45, 1211–1220.
- Fagan, V.; Bonham, S.; Carty, M.P.; Aldabbagh, F. One-pot double intramolecular homolytic aromatic substitution routes to dialicyclic ring fused imidazobenzimidazolequinones and preliminary analysis of anticancer activity. Org. Biomol. Chem. 2010, 8, 3149–3156.
- Fagan, V.; Bonham, S.; McArdle, P.; Carty, M.P.; Aldabbagh, F. Synthesis and toxicity of new ring-fused imidazo[5,4-f]benzimidazolequinones and mechanism using amine N-oxide cyclizations. Eur. J. Org. Chem. 2012, 2012, 1967–1975.
- Fagan, V.; Bonham, S.; Carty, M.P.; Saenz-Méndez, P.; Eriksson, L.A.; Aldabbagh, F. COMPARE analysis of the toxicity of an iminoquinone derivative of the imidazo[5,4-f]benzimidazoles with NAD(P)H:quinone oxidoreductase 1 (NQO1) activity and computational docking of quinones as NQO1 substrates. Bioorg. Med. Chem. 2012, 20, 3223–3232.
- Conboy, D.; Aldabbagh, F. 6-Imino-1,2,3,4,8,9,10,11-octahydropyrido[1,2-a]pyrido[1′,2′:1,2]imidazo[4,5-f]benzimidazole-13-one: Synthesis and cytotoxicity evaluation. Molbank 2020, 2020, M118.
- Sharma, A.; Arambula, J.F.; Koo, S.; Kumar, R.; Singh, H.; Sessler, J.L.; Kim, J.S. Hypoxia-targeted drug delivery. Chem. Soc. Rev. 2019, 48, 771–813.
- Zhang, K.; Chen, D.; Ma, K.; Wu, X.; Hao, H.; Jiang, S. NAD(P)H:Quinone Oxidoreductase 1 (NQO1) as a therapeutic and diagnostic target in cancer. J. Med. Chem. 2018, 61, 6983–7003.
- Dawood, K.M.; Abdel-Wahab, B.F. Synthetic routes to benzimidazole-based fused polyheterocycles. ARKIVOC 2010, 333–389.
- Dawood, K.M.; Elwan, N.M.; Abdel-Wahab, B.F. Recent advances on the synthesis of azoles, azines and azepines fused to benzimidazole. ARKIVOC 2011, 111–195.
- Khajuria, R.; Rasheed, S.; Khajuria, C.; Kapoor, K.K.; Das, P. Recent developments in the synthesis of pyrido[1,2-a]benzimidazoles. Synthesis 2018, 50, 2131–2149.
- Manna, S.K.; Das, T.; Samanta, S. Polycyclic benzimidazole: Synthesis and photophysical properties. ChemistrySelect 2019, 4, 8781–8790.
- Spiegel, L.; Kaufmann, H. Reduction of dinitrophenylpiperidine. II. Communication. Ber. Dtsch. Chem. Ges. 1908, 41, 679–685.
- Bamberger, E.; Tschirner, F. Direct transformation of the aniline in phenylhydroxylamine. Ber. Dtsch. Chem. Ges. 1899, 32, 1675–1678.
- Nair, M.D.; Adams, R. Benzimidazole syntheses by oxidative cyclization with peroxytrifluoroacetic acid. J. Am. Chem. Soc. 1961, 83, 3518–3521.
- Meth-Cohn, O.; Suschitzky, H. Syntheses of heterocyclic compounds. Part IV. Oxidative cyclisation of aromatic amines and their N-acyl derivatives. J. Chem. Soc. 1963, 4666–4669.
- Meth-Cohn, O. Mechanism of formation of benzimidazoles by oxidation of o-acylamino-N,N-dialkylanilines with peroxy-acids. J. Chem. Soc. C 1971, 1356–1357.
- Fahey, K.; Aldabbagh, F. Synthesis of seven- and eight-membered [1,2-a] alicyclic ring-fused benzimidazoles and 3-aziridinylazepino[1,2-a]benzimidazolequinone as a potential antitumour agent. Tetrahedron Lett. 2008, 49, 5235–5237.
- Gurry, M.; McArdle, P.; Aldabbagh, F. Synthesis of a spirocyclic oxetane-fused benzimidazole. Molecules 2015, 20, 13864–13874.
- Hussain, H.; Green, I.R.; Ahmed, I. Journey describing applications of Oxone in synthetic chemistry. Chem. Rev. 2013, 113, 3329–3371.
- Alkhader, M.A.; Perera, R.C.; Sinha, R.P.; Smalley, R.K. Synthesis of polynuclear heterocycles. Part 4.l imidazo[4,5-g] [3,1]-benzoxazinones, imidazo[4,5-g]quinazolinones, imidazo[4,5-g]quinazolinediones, and imidazo[4,5-f]indazolinones. J. Chem. Soc. Perkin 1 1979, 1056–1062.
- Sweeney, M.; Gurry, M.; Keane, L.-A.J.; Aldabbagh, F. Greener synthesis using hydrogen peroxide in ethyl acetate of alicyclic ring-fused benzimidazoles and anti-tumour benzimidazolequinones. Tetrahedron Lett. 2017, 58, 3565–3567.
- Gernon, M.D.; Wu, M.; Buszta, T.; Janney, P. Environmental benefits of methanesulfonic acid. Comparative properties and advantages. Green Chem. 1999, 1, 127–140.
- Meth-Cohn, O.; Suschitzky, H.; Sutton, M.E. Oxidative cyclisations of o-substituted anilines and benzoic acids with manganese dioxide. J. Chem. Soc. C 1968, 1722–1726.
- Möhrle, H. Gerloff, Tricyclische benzimidazolderivate. J. Arch. Pharm. 1978, 311, 381–393.
- Sun, X.; Hu, Y.; Nie, S.-Z.; Yan, Y.-Y.; Zhang, X.-J.; Yan, M. Efficient construction of C=N double bonds via acceptorless dehydrogenative coupling. Adv. Synth. Catal. 2013, 355, 2179–2184.
- Sun, X.; Lv, X.-H.; Ye, L.-M.; Hu, Y.; Chen, Y.-Y.; Zhang, X.-J.; Yan, M. Synthesis of benzimidazoles via iridium-catalyzed acceptorless dehydrogenative coupling. Org. Biomol. Chem. 2015, 13, 7381–7383.
- Xue, D.; Long, Y.-Q. Metal-free TEMPO-promoted C(sp3)−H amination to afford multisubstituted benzimidazoles. J. Org. Chem. 2014, 79, 4727–4734.
- Thapa, P.; Palacios, P.M.; Tran, T.; Pierce, B.S.; Foss, F.W., Jr. 1,2-Disubstituted benzimidazoles by the iron catalyzed cross-dehydrogenative coupling of isomeric o-phenylenediamine substrates. J. Org. Chem. 2020, 85, 1991–2009.
- Li, Q.-Y.; Cheng, S.-Y.; Tang, H.-T.; Pan, Y.-M. Synthesis of rutaecarpine alkaloids via an electrochemical cross dehydrogenation coupling reaction. Green Chem. 2019, 21, 5517–5520.
- Begunov, R.S.; Sakulina, V.O.; Syroeshkin, M.A.; Saverina, E.A.; Sokolov, A.A.; Minyaev, M.E. Electroreductive heterocyclization of ortho-piperidino substituted nitro(het)arenes. Mendeleev Commun. 2020, 30, 633–635.
- Martin, J.; Meth-Cohn, O.; Suschitzky, H. A simple route to polychlorobenzimidazoles and related systems. Tetrahedron Lett. 1973, 14, 4495–4498.
- Gurry, M.; Sweeney, M.; McArdle, P.; Aldabbagh, F. One-pot hydrogen peroxide and hydrohalic acid induced ring closure and selective aromatic halogenation to give new ring-fused benzimidazoles. Org. Lett. 2015, 17, 2856–2859.
- Sweeney, M.; Keane, L.-A.J.; Gurry, M.; McArdle, P.; Aldabbagh, F. One-pot synthesis of dihalogenated ring-fused benzimidazolequinones from 3,6-dimethoxy-2-(cycloamino)anilines using hydrogen peroxide and hydrohalic acid. Org. Lett. 2018, 20, 6970–6974.
- Conboy, D.; Mirallai, S.I.; Craig, A.; McArdle, P.; Al-Kinani, A.A.; Barton, S.; Aldabbagh, F. Incorporating morpholine and oxetane into benzimidazolequinone antitumor agents: The discovery of 1,4,6,9-tetramethoxyphenazine from hydrogen peroxide and hydroiodic acid-mediated oxidative cyclizations. J. Org. Chem. 2019, 84, 9811–9818.
- Parr, R.G.; Szentpály, L.v.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924.
- Meth-Cohn, O.; Suschitzky, H. Heterocycles by ring closure of ortho-substituted t-anilines (The t-amino effect). Adv. Heterocycl. Chem. 1972, 14, 211–278.
- Kwast, A.; Stachowska, K.; Trawczyński, A.; Wróbel, Z. N-Aryl-2-nitrosoanilines as intermediates in the synthesis of substituted phenazines from nitroarenes. Tetrahedron Lett. 2011, 52, 6484–6488.
- Guttenberger, N.; Blankenfeldt, W.; Breinbauer, R. Recent developments in the isolation, biological function, biosynthesis, and synthesis of phenazine natural products. Bioorg. Med. Chem. 2017, 25, 6149–6166.
- Conboy, D.; Aldabbagh, F. The reactivity of Oxone towards 4,6-di(cycloamino)-1,3-phenylenediamines: Synthesis of spirocyclic oxetane ring-fused imidazobenzimidazoles. ARKIVOC 2020, 180–191.
- Purkait, A.; Roy, S.K.; Srivastava, H.K.; Jana, C.K. Metal-free sequential C(sp2)−H/OH and C(sp3)−H aminations of nitrosoarenes and N-heterocycles to ring-fused imidazoles. Org. Lett. 2017, 19, 2540–2543.