The transient receptor potential melastatin (TRPM) family belongs to the superfamily of TRP ion channels. It consists of eight family members that are involved in a plethora of cellular functions. TRPM2 is a homotetrameric Ca2+-permeable cation channel activated upon oxidative stress and is important, among others, for body heat control, immune cell activation and insulin secretion. Invertebrate TRPM2 proteins are channel enzymes; they hydrolyze the activating ligand, ADP-ribose, which is likely important for functional regulation. Since its cloning in 1998, the understanding of the biophysical properties of the channel has greatly advanced due to a vast number of structure–function studies. The physiological regulators of the channel have been identified and characterized in cell-free systems. In the wake of the recent structural biochemistry revolution, several TRPM2 cryo-EM structures have been published. These structures have helped to understand the general features of the channel, but at the same time have revealed unexplained mechanistic differences among channel orthologues.
- TRPM2
- ion channels
- single particle cryo-EM
- ADP-ribose
- Nudix hydrolase
1. Introduction
The transient receptor potential (TRP) superfamily is a large cluster of transmembrane proteins consisting of ion channels that are expressed in a large variety of tissues. The superfamily falls into several families: canonical TRPC (1–7), vanilloid TRPV (1–6), vanilloid-like TRPVL, melastatin-like TRPM (1–8), polycystin TRPP (2, 3 and 5), mucolipin TRPML (1–3), ankyrin-rich TRPA (7 subfamilies), soromelastatin TRPS and no mechanoreceptor potential C TRPN (or NOMPC) [1]. TRP channels sense a plethora of signals, including ion concentration changes in the cell, binding of lipids or other ligands, temperature changes and mechanical signals, and respond with pore opening and transmembrane cation flux [2]. The largest family is TRPM, which has eight members. Most TRPM channels conduct both mono- and divalent cations, while TRPM4 and 5 are Ca
TRPM2 channels are widely expressed in bone marrow, the heart, liver, pancreas, leukocytes, lungs, spleen, eye, and brain [5][6][7]. TRPM2 is a nonselective cation channel which is co-activated by ADP-ribose (ADPR) and Ca
. TRPM2 activity is indispensable for many physiological processes including insulin secretion in pancreatic β-cells [8], monocyte chemokine production [9] and heat sensation of hypothalamic neurons [10]. Due to its permeability to Ca
, TRPM2 is also involved in many pathophysiological processes that lead to cell death [11], including neuronal cell death upon reactive oxygen species (ROS) production and in neurodegenerative disorders [5][12][13][14]. Hence, TRPM2 has become an attractive pharmacological target.
TRPM2 is a homotetramer; its subunits are built from ~1500 amino acid residues and consist of a large cytosolic N-terminal region, followed by a transmembrane domain (TMD) and a cytosolic C-terminal region. Both the intracellular and transmembrane regions are involved in ligand binding. The TMD contains six transmembrane helices (S1–6), and its structural organization resembles that of voltage-dependent cation channels [15][16]. In voltage-gated K
channels S1–S4 constitute the voltage sensor domain (VSD) which harbors four conserved arginines that move in response to voltage changes (the “gating charge”), causing strong voltage sensitivity of channel gating. In the TRPM family, the overall structural organization of the VSD is conserved, but the four arginines are absent. Correspondingly, TRPM4, 5 and 8 are weakly voltage sensitive, while TRPM2 is largely voltage insensitive. The S5–S6 region forms the pore domain (PD) which embraces an extracellular non-selective cation permissive selectivity filter and a cytoplasmic gate that regulates channel open probability. TRPM2 is unique among other family members in that its C-terminal region is extended by an ~270 amino acid-long cytosolic domain which shares ~40% sequence homology with the mitochondrial NUDT9 protein [7] and is hence called NUDT9-homology (NUDT9-H) domain. The mitochondrial NUDT9 enzyme belongs to the group of nucleoside diphosphate-linked moiety X (Nudix) hydrolases, which all contain a canonical Nudix motif sequence, GX
EX
REUXEEXGU, where X refers to any, while U to a large hydrophobic residue [17]. The Nudix motif, and especially the Nudix box (REUXEE), is thought to be important for the stability of the active site and coordination of cationic cofactors. Mutations introduced into canonical Nudix-box positions abolish hydrolytic activity of Nudix hydrolases, as shown, e.g., for NUDT9 by RILGEE and REFGKK substitutions of its Nudix box (REFGEE) [18]. The mechanism of the enzymatic reaction is general base catalysis. In pyrophosphatases, such as NUDT9, a water-driven nucleophilic attack on the alpha phosphorus initiates hydrolysis and is facilitated by divalent cation cofactors, typically Mg
. The nature of the catalytic base is variable, either a glutamate within the Nudix box [19] or a glutamate [20] or histidine outside the Nudix sequence [21]. NUDT9 is a specific ADP-ribose (ADPR) pyrophosphatase (ADPRase), which cleaves ADPR into AMP and ribose-5-phosphate. An N-terminal subdomain (cap), consisting of β-hairpins and α-helices, caps the C-terminal subdomain (core). The core subdomain, which consists of several α-helices and pleated β-sheets, harbors the catalytic site, and shows the highest homology with other Nudix hydrolases [22]. While the isolated core subdomain retains activity and specificity for ADPR, the cap subdomain is also involved in correct substrate positioning [18]. ADPR supposedly binds into a cleft between the core and the cap subdomain, where its hydrolysis product ribose-5-phosphate was detected in structures [22].
In cells, ADPR is produced from several metabolic pathways. Sir2 (class III protein deacetylase) enzymes catalyze the removal of lysine-bound acetyl groups and use NAD as a cofactor. One reaction end product is 2’- or 3’-O-acetyl-ADPR (OAADPR), which is converted into ADPR by several cytosolic enzymes [23]. Interestingly, OAADPR itself was reported to bind to the NUDT9-H domain of human TRPM2 (hsTRPM2) with a similar affinity as ADPR [24], while mitochondrial NUDT9 binds OAADPR with a 500-fold lower affinity than ADPR. ROS generation in either immune cells or in the brain upon ischemic stroke elevates cytosolic ADPR, leading to Ca
entry into cells through TRPM2 channels. ROS, on the one hand, induce the activation of the DNA repair enzymes poly-ADPR polymerase and glycohydrolase, which produce cytosolic ADPR. On the other hand, the ADPR precursor NAD
and possibly ADPR itself might be released from the mitochondrial matrix through the mitochondrial permeability transition pore [11].
2. Characterization of TRPM2 in a Cell-Free System and Identification of Direct Effectors
2.1. Activation of TRPM2 by ADPR, Ca2+ and Phosphatidylinositol-4,5-bisphosphate (PIP2)
Soon after cloning of the TRPM2 channel [25], whole-cell patch-clamp studies showed that the channel is activated by intracellular ADPR and activation requires Ca
[7][26][27][28][29]. As in whole-cell studies the composition of the cytosolic solution, and particularly that of microdomains, is not strictly controlled, the location of the activating Ca
binding sites (extra- or intracellular) could not be unambiguously determined [27][29]. In contrast, inside-out patch clamp recordings afford strict control over the composition of the cytosolic solution, and rapid application/removal (on the timescale of tens of milliseconds) of intracellular ligands. In patches excised from
oocytes injected with hsTRPM2 cRNA, large TRPM2 currents were evoked by simultaneous application of ADPR and Ca
(
A) [30][31]. Both ligands were required for channel activity, but interestingly, effective concentrations differed from those published earlier in studies using whole cells [7][24][26][27][28][29]. Whereas in whole-cell recordings EC
for ADPR and Ca
were ~100 μM and ~300 nM, respectively [7][27], in inside-out patches, ADPR apparent affinity was two orders of magnitude higher (EC
~1 μM) whilst that of Ca
was two orders of magnitude lower (EC
~20 μM), and these apparent affinities were little affected by the concentration of the other ligand [30][31]. A possible explanation for a lower Ca
affinity could have been the loss of calmodulin in the patch (cf [29][32]); however, the addition of external bovine calmodulin had no effect on fractional currents at micromolar Ca
concentrations [31]. Since even 1 mM ADPR elicited only minimal activation in submicromolar Ca
, the TRPM2 channel seems to be intrinsically set to sense high local Ca
concentrations.
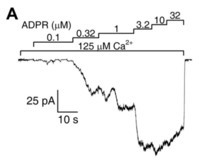
Functional characterization of TRPM2. (
) Representative trace of a macroscopic inside-out patch-clamp measurement. Downward deflections reflect channel opening and flux of Na
ions in response to indicated stimulating ADPR and Ca
concentrations. Scaling of current and time are indicated (L-bar). Figure is adapted from [31].
While no structural data were available at the time, functional observations in inside-out patches allowed rough localization of the Ca
binding sites. In steady-state single channel recordings, channel closed-time durations were independent of extracellular Ca
concentration, suggesting that in closed channels the binding sites are shielded from extracellular Ca
[30]. On the other hand, macroscopic channel closure upon sudden intracellular Ca
withdrawal could be incrementally slowed by raising extracellular Ca
[30], suggesting that as long as the pore is open extracellular Ca
can reach its binding sites despite Ca
-free vectorial rinsing of the cytosolic channel surface. These findings strongly suggested that the Ca
binding sites are located in an intracellular crevice very near the channel pore.
Besides the two obligate ligands, ADPR and Ca
, the presence of membrane phosphatidylinositol-4,5-bisphosphate (PIP
) is also a prerequisite for channel activation, as in inside-out patches masking the negatively charged headgroups of endogenous PIP
with the polycation polylysine shuts TRPM2 pores, which can then be re-opened only by millimolar amounts of Ca
[33] or by administration with external PIP
[34]. While H
O
potently stimulates TRPM2 currents in intact cells [5][26][35], this effect must be a secondary consequence of ADPR and/or Ca
release in response to the induced oxidative stress [35], as H
O
itself was ruled out as a direct effector of TRPM2 channel activity in cell-free recordings [31].
2.2. Pyridine Nucleotides and Their Derivatives Are Not Direct Activators of TRPM2
In the body, CD38, a multifunctional glycoprotein enzyme present on the surface of immune cells, converts the pyridine nucleotides NAD and NAAD into ADPR, but NADP and NAADP into ADPR-2-phosphate (ADPRP) [36]. These nucleotides and others, the metabolism of which is intertwined with that of ADPR, were tested to address the potential effects on TRPM2. NAD was identified as a channel agonist in early studies [5][28], while no binding to the NUDT9-H domain was later reported [24]. NAAD [31], NAADP [31][37][38] and cADPR [37][38] were reported to act as activators, AMP as an inhibitor [24][26], while NADH and NADP were found to be ineffective [5]. Besides these reported effects on channel activity, cADPR [26][37][38] and NAADP [37][38] were also shown to augment the effects of ADPR in a positively cooperative manner. 8-Br-cADPR, an agent shown to reduce ischæmic acute kidney injury [37], was suggested to act as a partial TRPM2 agonist and a competitive inhibitor of activation by cADPR [26]. Most of the above studies examined the effects of nucleotides in whole-cell systems. In a study in inside-out patches, the effects of cADPR on TRPM2 channel activity were shown to be attributable to ADPR contamination [31]. Decontamination of commercial cADPR stocks with a nucleotide pyrophosphatase which selectively cleaves ADPR but not cADPR eliminated channel stimulation by the compound in excised patches. Furthermore, no cooperative effects of cADPR with ADPR were observed. Recently, Yu et al reported that synthetic pure cADPR directly binds to the human NUDT9-H domain [39]. SPR titration indicated an apparent affinity (K
) of ~12 μM; however, much higher concentrations of cADPR (EC
~250 μM) were required to stimulate macroscopic TRPM2 currents. Considering its submicromolar cellular concentration [40], cADPR does not seem to act as a physiological activator of TRPM2. TRPM2 channel activation by various pyridine dinucleotides is explained by their spontaneous degradation, which also produces ADPR(P). Cleavage of contaminating ADPR in NAD and NAAD stocks, or of contaminating ADPRP in NAADP stocks, by the purified NUDT9 ADPRase abolished the stimulatory effects of the dinucleotides, ruling them out as direct effectors of TRPM2 [41]. Moreover, these experiments also demonstrated that none of the spontaneous and/or enzymatic cleavage products—nicotinamide, nicotinic acid, AMP(P) or ribose-5-phosphate—are TRPM2 channel activators. An apparent stimulatory effect of NAADP prior to decontamination pinpointed its spontaneous degradation product ADPRP as an agonist. Indeed, in inside-out patches, pure ADPRP opened TRPM2 channels, albeit its apparent affinity was lower (EC
~13 μM) and maximal stimulation smaller (~80%) when compared to ADPR. The lower open probability supported by ADPRP was explained by an approximately three-times faster macroscopic closing rate, reflecting a shortened open burst [41]. A similar efficacy but even lower affinity (EC
~110 μM) for ADPRP was reported in a whole-cell study [42] that also showed 2’-deoxy-ADPR (dADPR) to act as a superagonist on hsTRPM2 channels. In the presence of dADPR, whole-cell TRPM2 currents were 37% larger, and following patch excision channels inactivated slower, but the EC
of dADPR was similar to that of ADPR. Since upon oxidative stress-induced DNA damage, dADPR may accumulate due to CD38 activity, this nucleotide is a likely regulator of TRPM2 function under pathophysiological conditions.
In summary, recordings in cell-free systems demonstrated that (d)ADPR is a physiologically relevant activator of the hsTRPM2 channel and ADPRP is a partial agonist, whereas AMP and pyridine dinucleotides are without effect [31][41] and cADPR is probably a non-physiological low affinity agonist [39]. Note, however, that nucleotide affinity and efficacy profiles differ among channel orthologues.
References
- Wang, H.; Siemens, J. TRP ion channels in thermosensation, thermoregulation and metabolism. Temperature 2015, 2, 178–187.
- Huang, Y.; Fliegert, R.; Guse, A.H.; Lü, W.; Du, J. A structural overview of the ion channels of the TRPM family. Cell Calcium 2020, 85, 102111.
- Launay, P.; Fleig, A.; Perraud, A.-L.; Scharenberg, A.M.; Penner, R.; Kinet, J.-P. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell 2002, 109, 397–407.
- Liu, D.; Liman, E.R. Intracellular Ca2+ and the phospholipid PIP2 regulate the taste transduction ion channel TRPM5. Proc. Natl. Acad. Sci. USA 2003, 100, 15160–15165.
- Hara, Y.; Wakamori, M.; Ishii, M.; Maeno, E.; Nishida, M.; Yoshida, T.; Yamada, H.; Shimizu, S.; Mori, E.; Kudoh, J.; et al. LTRPC2 Ca2+-Permeable Channel Activated by Changes in Redox Status Confers Susceptibility to Cell Death. Mol. Cell 2002, 9, 163–173.
- Kühn, F.J. Structure-Function Relationship of TRPM2: Recent Advances, Contradictions, and Open Questions. Int. J. Mol. Sci. 2020, 21, 6481.
- Perraud, A.-L.; Fleig, A.; Dunn, C.A.; Bagley, L.A.; Launay, P.; Schmitz, C.; Stokes, A.J.; Zhu, Q.; Bessman, M.J.; Penner, R.; et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nat. Cell Biol. 2001, 411, 595–599.
- Uchida, K.; Tominaga, M. The role of thermosensitive TRP (transient receptor potential) channels in insulin secretion. Endocr. J. 2011, 58, 1021–1028.
- Yamamoto, S.; Shimizu, S.; Kiyonaka, S.; Takahashi, N.; Wajima, T.; Hara, Y.; Negoro, T.; Hiroi, T.; Kiuchi, Y.; Okada, T.; et al. TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflam-matory neutrophil infiltration. Nat. Med. 2008, 14, 738–747.
- Song, K.; Wang, H.; Kamm, G.B.; Pohle, J.; Reis, F.D.C.; Heppenstall, P.; Wende, H.; Siemens, J. The TRPM2 channel is a hypothalamic heat sensor that limits fever and can drive hypothermia. Science 2016, 353, 1393–1398.
- Nilius, B.; Owsianik, G.; Voets, T.; Peters, J.A. Transient Receptor Potential Cation Channels in Disease. Physiol. Rev. 2007, 87, 165–217.
- Fonfria, E.; Marshall, I.C.B.; Boyfield, I.; Skaper, S.D.; Hughes, J.P.; E Owen, D.; Zhang, W.; Miller, B.A.; Benham, C.D.; McNulty, E.S. Amyloid beta-peptide (1–42) and hydrogen peroxide-induced toxicity are mediated by TRPM2 in rat primary striatal cultures. J. Neurochem. 2005, 95, 715–723.
- Hermosura, M.C.; Cui, A.M.; Go, R.C.V.; Davenport, B.; Shetler, C.M.; Heizer, J.W.; Schmitz, C.; Mocz, G.; Garruto, R.M.; Perraud, A.-L. Altered functional properties of a TRPM2 variant in Guamanian ALS and PD. Proc. Natl. Acad. Sci. USA 2008, 105, 18029–18034.
- Kaneko, S.; Kawakami, S.; Hara, Y.; Wakamori, M.; Itoh, E.; Minami, T.; Takada, Y.; Kume, T.; Katsuki, H.; Mori, Y.; et al. A Critical Role of TRPM2 in Neuronal Cell Death by Hydrogen Peroxide. J. Pharmacol. Sci. 2006, 101, 66–76.
- Long, S.B.; Campbell, E.B.; MacKinnon, R. Crystal Structure of a Mammalian Voltage-Dependent Shaker Family K+ Channel. Science 2005, 309, 897–903.
- Long, S.B.; Campbell, E.B.; MacKinnon, R. Voltage Sensor of Kv1.2: Structural Basis of Electromechanical Coupling. Science 2005, 309, 903–908.
- Mildvan, A.; Xia, Z.; Azurmendi, H.; Saraswat, V.; Legler, P.; Massiah, M.; Gabelli, S.; Bianchet, M.; Kang, L.-W.; Amzel, L. Structures and mechanisms of Nudix hydrolases. Arch. Biochem. Biophys. 2005, 433, 129–143.
- Perraud, A.-L.; Shen, B.; Dunn, C.A.; Rippe, K.; Smith, M.K.; Bessman, M.J.; Stoddard, B.L.; Scharenberg, A.M. NUDT9, a Member of the Nudix Hydrolase Family, Is an Evolutionarily Conserved Mitochondrial ADP-ribose Pyrophosphatase. J. Biol. Chem. 2003, 278, 1794–1801.
- Harris, T.K.; Wu, G.; Massiah, M.A.; Mildvan, A.S. Mutational, Kinetic, and NMR Studies of the Roles of Conserved Glutamate Residues and of Lysine-39 in the Mechanism of the MutT Pyrophosphohydrolase†. Biochemistry 2000, 39, 1655–1674.
- Gabelli, S.B.; Bianchet, A.M.; Ohnishi, Y.; Ichikawa, Y.; Bessman, M.J.; Amzel, L.M. Mechanism of the Escherichia coli ADP-ribose pyrophosphatase, a Nudix hydrolase. Biochemistry 2002, 41, 9279–9285.
- Legler, P.M.; Massiah, M.A.; Mildvan, A.S. Mutational, Kinetic, and NMR Studies of the Mechanism ofE. coliGDP-Mannose Mannosyl Hydrolase, an Unusual Nudix Enzyme†. Biochemistry 2002, 41, 10834–10848.
- Shen, B.W.; Perraud, A.L.; Scharenberg, A.; Stoddard, B.L. The Crystal Structure and Mutational Analysis of Human NUDT9. J. Mol. Biol. 2003, 332, 385–398.
- Tong, L.; Denu, J.M. Function and metabolism of sirtuin metabolite O-acetyl-ADP-ribose. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2010, 1804, 1617–1625.
- Grubisha, O.; Rafty, L.A.; Takanishi, C.L.; Xu, X.; Tong, L.; Perraud, A.-L.; Scharenberg, A.M.; Denu, J.M. Metabolite of SIR2 Reaction Modulates TRPM2 Ion Channel. J. Biol. Chem. 2006, 281, 14057–14065.
- Nagamine, K.; Kudoh, J.; Minoshima, S.; Kawasaki, K.; Asakawa, S.; Ito, F.; Shimizu, N. Molecular cloning of a novel putative Ca2+ channel protein (TRPC7) highly expressed in brain. Genomics 1998, 54, 124–131.
- Kolisek, M.; Beck, A.; Fleig, A.; Penner, R. Cyclic ADP-Ribose and Hydrogen Peroxide Synergize with ADP-Ribose in the Activation of TRPM2 Channels. Mol. Cell 2005, 18, 61–69.
- McHugh, D.; Flemming, R.; Xu, S.-Z.; Perraud, A.-L.; Beech, D.J. Critical Intracellular Ca2+ Dependence of Transient Receptor Potential Melastatin 2 (TRPM2) Cation Channel Activation. J. Biol. Chem. 2003, 278, 11002–11006.
- Sano, Y.; Inamura, K.; Miyake, A.; Mochizuki, S.; Yokoi, H.; Matsushime, H.; Furuichi, K. Immunocyte Ca2+ Influx System Mediated by LTRPC2. Science 2001, 293, 1327–1330.
- Starkus, J.; Beck, A.; Fleig, A.; Penner, R. Regulation of TRPM2 by Extra- and Intracellular Calcium. J. Gen. Physiol. 2007, 130, 427–440.
- Csanady, L.; Torocsik, B. Four Ca2+ ions activate TRPM2 channels by binding in deep crevices near the pore but in-tracellularly of the gate. J. Gen Physiol. 2009, 133, 189–203.
- Tóth, B.; Csanády, L. Identification of Direct and Indirect Effectors of the Transient Receptor Potential Melastatin 2 (TRPM2) Cation Channel*. J. Biol. Chem. 2010, 285, 30091–30102.
- Tong, Q.; Zhang, W.; Conrad, K.; Mostoller, K.; Cheung, J.Y.; Peterson, B.Z.; Miller, B.A. Regulation of the Transient Receptor Potential Channel TRPM2 by the Ca2+ Sensor Calmodulin. J. Biol. Chem. 2006, 281, 9076–9085.
- Tóth, B.; Csanády, L. Pore collapse underlies irreversible inactivation of TRPM2 cation channel currents. Proc. Natl. Acad. Sci. USA 2012, 109, 13440–13445.
- Zhang, Z.; Tóth, B.; Szollosi, A.; Chen, J.; Csanády, L. Structure of a TRPM2 channel in complex with Ca2+ explains unique gating regulation. eLife 2018, 7, e36409.
- Perraud, A.-L.; Takanishi, C.L.; Shen, B.; Kang, S.; Smith, M.K.; Schmitz, C.; Knowles, H.M.; Ferraris, D.; Li, W.; Zhang, J.; et al. Accumulation of Free ADP-ribose from Mitochondria Mediates Oxidative Stress-induced Gating of TRPM2 Cation Channels. J. Biol. Chem. 2005, 280, 6138–6148.
- Kim, H.; Jacobson, E.L.; Jacobson, M.K. Synthesis and degradation of cyclic ADP-ribose by NAD glycohydrolases. Science 1993, 261, 1330–1333.
- Beck, A.; Kolisek, M.; Bagley, L.A.; Fleig, A.; Penner, R. Nicotinic acid adenine dinucleotide phosphate and cyclic ADP-ribose regulate TRPM2 channels in T lymphocytes. FASEB J. 2006, 20, 962–964.
- Lange, I.; Penner, R.; Fleig, A.; Beck, A. Synergistic regulation of endogenous TRPM2 channels by adenine dinucleotides in primary human neu-trophils. Cell Calcium 2008, 44, 604–615.
- Yu, P.; Liu, Z.; Yu, X.; Ye, P.; Liu, H.; Xue, X.; Yang, L.; Li, Z.; Wu, Y.; Fang, C.; et al. Direct Gating of the TRPM2 Channel by cADPR via Specific Interactions with the ADPR Binding Pocket. Cell Rep. 2019, 27, 3684–3695.e4.
- Heiner, I.; Eisfeld, J.; Warnstedt, M.U.; Radukina, N.; Jüngling, E.; Lückhoff, A. Endogenous ADP-ribose enables calcium-regulated cation currents through TRPM2 channels in neutrophil granulocytes. Biochem. J. 2006, 398, 225–232.
- Tóth, B.; Iordanov, I.; Csanády, L. Ruling out pyridine dinucleotides as true TRPM2 channel activators reveals novel direct agonist ADP-ribose-2′-phosphate. J. Gen. Physiol. 2015, 145, 419–430.
- Fliegert, R.; Bauche, A.; Pérez, A.M.W.; Watt, J.M.; Rozewitz, M.D.; Winzer, R.; Janus, M.; Gu, F.; Rosche, A.; Harneit, A.; et al. 2’-Deoxyadenosine 5’-diphosphoribose is an endogenous TRPM2 superagonist. Nat. Chem. Biol. 2017, 13, 1036–1044.