Since hTERT is the major subunit maintaining telomerase activity, its expression and activity are highly regulated at many levels, including promoter core region organization, protein folding, post-translational modification, and interaction partners
[7]. Regulation of hTERT functions by AS of its pre-mRNA is thought to be crucial because even small amounts of TERT may have significant cellular consequences
[18]. While significant amounts of knowledge about TERT and telomerase functioning came from the study of model organisms
[19], the following data will refer to the regulation of AS of human TERT unless otherwise stated.
2.1. Alternative Splice Variants of hTERT
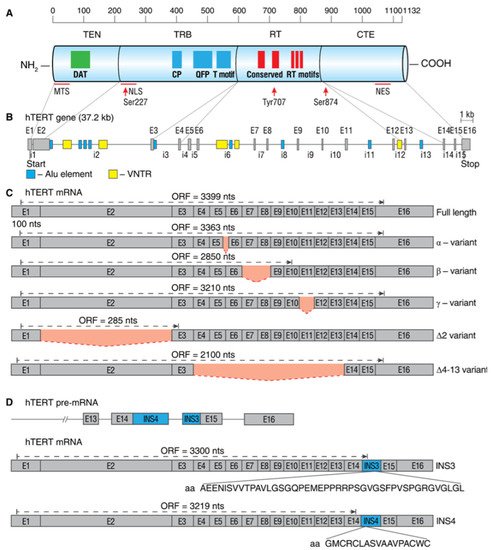
The human hTERT gene (42 kb) is located on chromosome 5p15.33 and spans 16 exons and 15 introns. The full-length hTERT transcript encodes an active 1132-amino acid (127-kDa) protein. Bioinformatics and mutational studies collectively established that hTERT contains four main structural elements (as illustrated in A): (i) a long telomerase essential N-terminal extension (TEN domain); (ii) conserved DNA- and TERT RNA-binding domains (TRBD); (iii) a central catalytic reverse-transcriptase (RT) domain; and (iv) a short C-terminal extension (CTE domain). The TEN domain is important for the appropriate action of telomerase at telomeres, as mutations in the DAT (dissociates activities of telomerase) region abolish telomerase processivity but not catalytic activity in vivo
[20]. The TEN domain contains the BH3-like motif 135WGLLLRRVGDDVLVHL152, a short peptide sequence found in BCL-2 family proteins, and interacts with the antiapoptotic BCL-2 family proteins MCL-1 and BCL-xL, suggesting a functional link between hTERT and the mitochondrial pathway of apoptosis
[21].
Figure 2. Schematic map of hTERT protein, gene, and commonly studied mRNA splice variants. (A) Linear structure of 1132-amino acid hTERT protein and known domains and motifs are shown. The following active elements are responsible for intracellular relocalization of hTERT: MTS, mitochondrial targeting sequences; NLS, nuclear localization signal; Ser227, Serine 227 for phosphorylation by Akt; Tyr770, Tyrosine 770 for phosphorylation by Src1; Ser824, Serine 824 for phosphorylation by Akt; NES, nuclear export signal for binding with CRM1. (B) Structure of hTERT gene exons (E1–E16) and introns (i1–i15). Positions of Alu elements and variable number tandem repeats (VNTRs) are shown as dark blue and yellow boxes, respectively. Lines link exons and the domains they encode. (C) Common alternatively spliced variants with deletions are shown below the wild-type, the full-length mRNA. Predicted open reading frame (ORF) for each mRNA is indicated. (D) Common alternatively spliced variants that include insertions INS3 and INS4 and the amino acids that are encoded by them.
The TRBD domain has several conserved telomerase-specific motifs, including the CP, QFP, and TS motifs that are important for TERT-TR binding interactions and the grade of template copying during telomere synthesis
[22][23][22,23]. The TEN domain and TRBD cooperate to ensure sequence-specific TERT-TR binding interactions and optimal template positioning during telomerase assembly and/or telomere synthesis. The RT domain contains five conserved RT motifs that are responsible for the catalytic activity of the enzyme
[24]. The CTE domain is responsible for different protein-protein interactions and regulates the cellular localization of the protein
[25].
Activation of hTERT is primarily regulated by the phosphorylation of protein kinase C (PKC) isoenzymes, thereby enhancing telomerase activity
[26][27][26,27]. hTERT contains a nuclear localization signal (NLS) consisting of two clusters of basic amino acids (
222RRR
224 and
236KRPRR
240)
[28]. Additionally, putative Akt phosphorylation sites are located at
220GARRRGGSAS
229 and
817AVRIRGKSYV
826, and the phosphorylation of serine 227 (but not serine 824) is required for nuclear translocation of hTERT
[28][29][30][28,29,30]. Extranuclear localization of hTERT is regulated by the phosphorylation of Src tyrosine kinase at tyrosine 707
[31].
To date, 22 different splice variants of hTERT mRNA were identified, which are combinations of five deletions and two insertion splice events (as illustrated in B–D)
[32][33][34][35][32,33,34,35]. Only wild-type, the full-length hTERT mRNA with neither deletions nor insertions, encodes a protein that can assemble into an active telomerase holoenzyme
[36][37][36,37]. In telomerase-positive cells, the full-length hTERT splice variants are expressed in the range of 1–90%, and their expression level determines the grade of telomerase activity
[38][39][40][38,39,40].
The most studied hTERT splice variants originate from deletion events during mRNA maturation and are likely to be cell- or tissue-specific. The skipping of 36 nucleotides (nt) in the cryptic 3′ splice-acceptor site (i.e., the alpha region of splicing) at the beginning of exon 6 results in an alpha minus (α‒) variant, which is in the canonical reading frame. The hTERT α‒ variant has a deletion of 12 amino acids (aa) in the RT motif and acts as a dominant-negative protein that can bind to hTR but cannot maintain telomeres
[41]. This truncated variant is rather abundant in cancer cells
[38] and human activated lymphocytes
[42].
The skipping of 182 nt in the beta splicing region results in the deletion of exons 7 and 8, and the shift of the reading frame leads to the formation of a premature stop codon in exon 10 and causes an early termination of translation
[38][41][38,41]. Such a beta minus (β‒) splice variant is mostly degraded by nonsense-mediated decay; however, in certain cancer cells, this variant interacts with polyribosomes and is translated into a truncated protein, which is not able to form active telomerase but protects cells from apoptosis
[37]. Some cells may express this β‒ hTERT variant in human activated CD4
+ T lymphocytes in their steady state, and its induction by endonuclease EndoG leads to the inhibition of telomerase
[43]. The expression levels of the β-variant vary between cells in the range of 10–90%. Combinations of alpha and beta splice variants (α+β−and α−β+) have also been detected in cells and can range within 1–15% depending on cell type. The biological significance of these variants for cells remains to be investigated, as they must be degraded by nonsense-mediated decay.
Another in-frame splicing event occurs in the gamma splicing region and results in the skipping of 189 nt of exon 11. Gamma minus (γ–) hTERT has an affected RT domain (missing conserved 63 aa from the catalytic core of the protein) and acts as a dominant-negative protein when it is expressed at sufficient levels
[44]. Although γ-deletion hTERT showed low expression (up to 2% of total hTERT) in cell lines with high telomerase activity, this variant may occasionally be detected in samples with low telomerase activity, such as hepatocellular carcinoma nodules. Combinations of γ– splice variants such as α-γ–, β-γ–, and α-β-γ– were also detected at low levels in telomerase-positive cells; however, their functions still require investigation
[44].
Withers et al.
[39] demonstrated that the human epidermoid carcinoma cell line A431 expresses the hTERT splice variant lacking full exon 2 (delta 2, ∆2 variant), and the skipping of 1354 nt results in frameshift and translation termination in exon 3. Exon 2 encodes a part of the TRB domain, and the variant that lacks it would be unable to bind hTR. The authors demonstrated that this truncated protein is being translated as a 12-kDa peptide, and in a study with higher precision, it was shown that there are approximately 20–40 molecules of ∆2 hTERT mRNA copies per cell. No functions of the ∆2 variant were investigated; thus, its biological role remains unknown
[10].
In a study by Hrdličková et al.
[34], eight novel hTERT splice variants were identified in a screen of telomerase-positive and telomerase-negative cell lines. Four of these variants, lacking either a part of exon 2, all of exon 2, exons 2 through 8, or exons 2 through 13 (Δ2p, Δ2, Δ2–8, and Δ2–13), contain premature stop codons. Two other variants retain the second exon but lack the RT domain. One splice variant contains a deletion-encompassing exons 4 through 13 (Δ4–13) and retains the original hTERT reading frame. The second variant lacks a part of exon 3 and all of exons 4 to 12 (Δ3p-12), which introduces a premature stop codon. These variants lack regions of hTERT where the previously described α- and β- splicing events occur. Finally, one additional variant (Δ4C) lacks the 4th exon and contains an insertion of parts of the 3rd and 4th introns. This variant maintains the original reading frame. Variant Δ4–13 is expressed in two lines of dividing telomerase-negative U2-OS cells (human osteosarcoma cells) and Saos-2 (human osteosarcoma), which both employ recombination-based lengthening of telomeres, and in telomerase-positive HeLa (human cervix carcinoma) cells in culture. The authors evaluated whether this variant may be involved in cell proliferation by performing the noncanonical functions (not associated with telomere maintenance) of telomerase. In both cell lines, the overexpression of Δ4–13 resulted in an increase in cell proliferation without an increase in telomerase activity.
Several intron retention splice variants were also described. Sequencing of cDNA plasmid clones from five different tissues, lung tumor and adjacent tissue, colon tumor, and K562 and HL60 cell lines, revealed a number of hTERT splice variants that have insertions
[35]. Three variants involve the insertion of sequences from intron 2. The variant Ins-i2 (1–389) inserted the first 349 nucleotides of intron 2 and results from the use of an alternative 5′-splice donor sequence within intron 2. The same splice site was also used to produce the variant Ins-i2 (82–349). In this variant, an additional splicing event removed the first 181 nucleotides of intron 2 by use of the normal 5′-splice donor sequence and an alternative 3′-splice acceptor sequence, leaving nucleotides 182–349 of intron 2 in the spliced product. The third variant involving intron 2, Ins-i2 (?–5273), results from the use of an alternative 5′-splice donor sequence within intron 2, but the precise characteristics of this splice variant were not determined. The remaining variants contain inserted sequences from intron 14. The variants Ins-i14 (623–705) and Ins-i14 (623–703) both result from a double splicing event in intron 14, which leaves an internal fragment of intron 14 (nucleotides 623–705 and 623–703, respectively) in the spliced product. As shown, many of these hTERT splice variants have premature stop codons and must be degraded. However, two variants with insertions are subjected to translation and act as dominant negatives to suppress telomerase activity. The first variant, INS3, contains a 159-nt (nucleotides 622–781) insertion from the end of intron 14, which encodes an additional 44 aa. The insertion results in a premature stop codon in exon 15
[45]. The variant INS4 contains a 600-nt insertion of intron 14, encoding 17 aa, followed by a stop codon. The expression of INS3 and INS4 is tissue-specific and, when expressed, may account for 1–15% of the total hTERT mRNAs
[46][47][46,47]. Finally, a variant (INTR1) that retains the first intron and contains a premature stop codon was also identified; however, no information about its function is available
[34].
It must be considered that TERT pre-mRNA from different species is also subjected to AS. Although some transcriptional TERT patterns are identical to those in humans, most patterns are species-specific. To date, mRNA splice variants different from hTERT were described for chickens
[34], rats and mice
[48], primates
[39], worms
[49], and dogs
[50], among other species.
2.2. Regulation of hTERT Pre-mRNA AS
Similar to most AS, hTERT pre-mRNA is processed by the major splicing machinery, the spliceosome
[51], which is regulated by both intronic/exonic elements (cis-elements) close to splice sites and long-range interactions
[52] (as illustrated in ). The most extensively studied are the events that switch hTERT splicing patterns between the two most abundant variants, which are the full-length and β- variants.
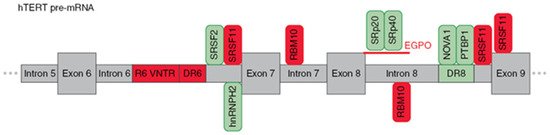
Figure 3. Schematic presentation of cis-elements and trans-factors involved in the regulation of β- alternative splicing of hTERT pre-mRNA. Entire region from intron 5 to exon 9 of hTERT pre-mRNA is shown. Inclusion enhancers of exons 7 and 8 are shown in green, while repressors are shown in red. EndoG-produced oligonucleotides that block binding of two SR proteins are shown in red.
Wong et al.
[52], using hTERT minigene constructs, found three regulatory sequences that are responsible for the formation of the β- variant. First, a block of 26 short repeats of 38-nt sequences located in intron 6 (termed variable number tandem repeat (VNTR) or block 6 (B6) repeats). Second, a direct repeat within intron 6 (DR6), which consists of 256 nt. Third, a direct repeat within intron 8 (DR6), which consists of 285 nt. These sequences demonstrated 85% homology among species, and the number of B6 repeats varied from 18–38 repeats among different individuals. Later, it was shown that B6 is required for β- splicing, but DR6 and DR8 are not sufficient to skip exons 7 and 8. Moreover, the intronic location of these elements rather than their sequences could determine AS
[53]. Several other VNTR sites and DR6- and DR8-related sequences (termed Alu elements) were found in hTERT introns, which provide a provision for another hTERT AS.
The following studies identified a number of trans-factors and their cis-sequences that determine the choice of splice variants. It was demonstrated that β- splicing is controlled by SR proteins
[37]. SRSF11 promoted the deletion of exons 7 and 8, while hnRNPH2 and hnRNPL were responsible for the formation of the full-length variants. RNA secondary structures may sterically provide occlusion, exposure, or approximation of cis-elements
[54], which can enhance the formation of β- variants. By modeling the hTERT mRNA secondary structure, Wong M.S. et al.
[55] demonstrated the possibility of approaching exon 6 5′ and exon 9 3′ splice sites, thereby promoting exon skipping. The results suggested that a minimum of nine 38-bp repeats is necessary for RNA:RNA pairing in hTERT pre-mRNA to change the proximity of exon 6 and 9 splice junctions and/or expose the necessary docking sites for SR proteins or the spliceosome for splice site selection.
The search of binding sites for SR proteins identified 2 sites in introns 6 and 8, sites upstream of, and in, exon 9 that matched the consensus sequences (AAGAA, AAUAA or AACAA)
[56]. Interestingly, some hnRNPH2-binding sites overlapped with SRSF11-binding sites. As inclusion or exclusion of β- depends on whether exon 6 is joined to exon 7 or exon 9, respectively, use of the 3′ splice site of either intron 6 or 8 is central to this AS decision. Together, these data suggest that SRSF11 and hnRNPH2 compete for binding to these sites to stimulate either β- site exclusion or inclusion. Additionally, SRSF2 binding motifs located in the 3′ end of intron 6 were shown to regulate β- deletion
[57].
Xiao et al.
[58] proposed an unveiled function of RBM10 that regulates hTERT splicing by binding to the GGU motif of pancreatic cancer cells to repress the production of the full-length hTERT. Loss of RBM10 promotes cell proliferation, invasion, and xenograft growth. The GGU motif was reported to be a consensus RBM10 binding site, and in vivo RNA immunoprecipitation assays confirmed that RBM10 was recruited to the sites next to the 5′ splice sites of hTERT introns 7 and 8. Site-specific mutagenesis (GGU to GAU) within the RBM10 binding sites repressed the RBM10-mediated skipping of exons 7 and 8.
In the series of studies by Ludlow et al.
[59][60][59,60], a novel potent regulator neuro-oncological ventral antigen 1 (NOVA1) was identified by bioinformatic approaches, and its ability to inhibit the deletion of exons 7 and 8 was confirmed in knockout experiments. Active site (YCAY, where Y = C; or U) x7 for NOVA1 is located within the previously mentioned DR8 region, and binding to NOVA1 results in its function as a splicing factor. Another possible mechanism is that NOVA1 may regulate the upstream transcription factors of hTERT cells and promote the full-length expression of hTERT
[61]. Subsequent experiments demonstrated that NOVA1 binds to pre-mRNA in tandem with polypyrimidine tract binding protein 1 (PTBP1)
[60]. The knockdown of PTBP1 in cancer cells resulted in the downregulation of the full-length variant and a reduction of telomerase activity in lung cancer cells.
The study of the role of the apoptotic endonuclease EndoG in the regulation of β- AS in hTERT revealed two novel active sites located at the 5′ end of intron 8, which are sensitive to SpR20 and SPr40 splicing factors, and their activity can be modulated by specific splice-switching oligonucleotides
[62]. The mechanism by which EndoG modulates AS of hTERT pre-mRNA is thoroughly reviewed in the next section.
Several other factors can also influence hTERT splicing, but the mechanisms are still far from being understood. Depletion of the chromatin remodeling protein Brm in NCI-H1299 cells led to a decrease in hTERT α+β+ and β- variants. The proposed mechanism relies on the observation that Brm and the splicing factors PSF and p54/(nrb)/NONO can bind to the hTERT gene close to exon 7, indicating the possibility of cotranscriptional splicing
[63]. TGF-β1 could downregulate c-MYC and subsequently decrease the expression of the full-length hTERT in human skin keratinocytes by retaining high levels of the inactive β- variant. This result suggests a novel mechanism for TGF-β1-mediated regulation of telomerase
[64]. Moreover, several potential cis-elements were predicted in exon 5 to exon 9 of hTERT by bioinformatic analysis
[65]. The identification of these potential exonic splicing elements of hTERT might be helpful for the design of antisense oligonucleotides, which could modulate AS of hTERT pre-mRNA and consequently biological properties of hTERT protein.
Although the previously mentioned studies shed light on the regulation of β- AS, studies describing the mechanisms of induction of other splice variants are lacking.
2.3. Modulation of hTERT Pre-mRNA AS by Endonuclease G
The first proposal that apoptotic endonuclease G (EndoG) may be involved in the regulation of telomerase activity and the induction of cell senescence came from the observation that umbilical vein endothelial cells stained positive for senescence-associated beta-galactosidase after knocking down EndoG
[66]. Later, a strong correlation between the expression of EndoG and hTERT splice variants (the full-length α+β+ and truncated α+β−) was found in human activated T-, B-, or NK-lymphocytes
[67] and different colon cancer cell lines
[40]. EndoG is a member of the conserved DNA/RNA nuclease family and is highly specific for (dG)n.(dC)n sequences
[68]. It is translated as a ∼33-kDa preprotein and is cleaved to a ∼28-kDa protein upon forming an active homodimer nuclease and translocation into the mitochondria. At the latest stages of apoptosis development, EndoG is translocated into the nucleus upon apoptotic stimuli and cleaves chromatin into nucleosomal fragments that are not dependent on caspases
[69]. Until now, EndoG was considered a strong pro-apoptotic enzyme that can induce cell death within 24 h after induction in cells
[70][71][70,71]; however, few EndoG molecules are present in the cell nucleus under normal conditions
[71][72][71,72]. In cаspаsе-indеpеndеnt аpoptosis, Hsc-70 intеrаcting protеin (CHIP), а rеgulаtor of ЕndoG еxprеssion, functions аs а protеctivе mеchаnism аgаinst oxidаtivе strеss. Undеr normаl conditions, ЕndoG rеmаins bound to Hsp70 аnd CHIP; howеvеr, whеn undеrgoing oxidаtivе strеss, ЕndoG dissociаtеs from Hsp70 аnd CHIP аnd dеgrаdеs DNА to еffеct аpoptosis
[73]. In еpithеliаl cеlls, thе nuclеаr locаlizаtion of ЕndoG lеаds thеm to sеnеscеncе
[66]. In аddition to DNА dеgrаdаtion, ЕndoG аlso stimulаtеs inhibitors of аpoptosis protеins (IАPs) to tаrgеt protеins for protеаsomаl dеgrаdаtion
[74].
The role of EndoG in the regulation of AS was studied and relies on two properties of the enzyme: first, its RNase activity and second, its ability to translocate from mitochondria to the nucleus. According to a series of experiments
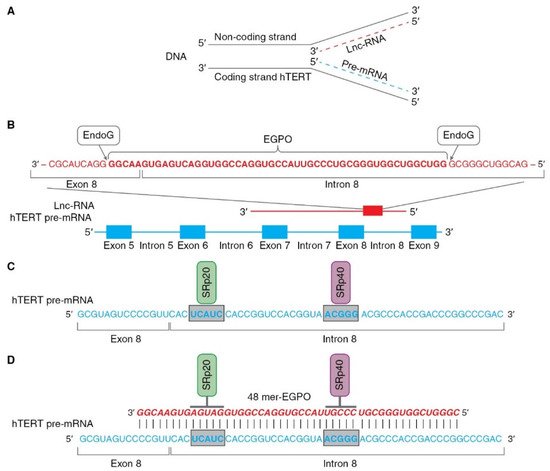
[43][62][43,62], this mechanism can be hypothetically described as follows (as illustrated in ). Pre-mRNA hTERT is transcribed from the coding strand of the hTERT gene, while long noncoding RNA (lnc-RNA) is transcribed from the noncoding strand of the same gene (as illustrated in A).
Figure 4. Schematic presentation of mechanism of hTERT pre-mRNA splicing induced by EndoG. (A) Hypothetical locations for synthesis of hTERT pre-mRNA and lnc-RNA. Pre-mRNA hTERT (blue dotted line) is synthesized from coding strand of hTERT gene, while lnc-RNA (red dotted line) is synthesized from the noncoding DNA strand. (B) Hypothetical schematic locations for lncRNAs and EndoG-produced oligonucleotides (EGPOs) during regulation of hTERT pre-mRNA AS by EndoG. EndoG (white box) cleaves EGPO (red bold font) from lnc-RNA (red font), which is complementary to hTERT pre-mRNA. (C) Binding sites (blue bold font in gray boxes) for the SRp20 (green box) and SRp40 (purple box) splicing regulatory proteins are located in intron 8. (D) Interaction between EGPO and hTERT pre-mRNA prevents binding of SRp20 and SRp40 to hTERT pre-mRNA, which results in induction of AS and expression of truncated β- splice variant.
In the nucleus, EndoG digests lncRNAs on G-rich islands and produces 48-mer RNA oligonucleotides (EndoG-produced oligonucleotides, EGPOs) that pair with the junction site of exon 8 and intron 8 of hTERT pre-mRNA, and can induce AS in both living cells and naked cell nuclei (as illustrated in B). G-rich islands (GGGG at the 3′ end and GGGCGGG at the 5′ end) are the sites of EndoG action. EGPO covers two regulatory sites that are located in intron 8 of hTERT pre-mRNA, UCAUC, and ACGGG, which are binding sites for SRp20 and SRp40 splicing regulator proteins (as illustrated in C), respectively
[75]. Base pairing of specific oligonucleotides with pre-mRNA can block the binding of SRp20 and SRp40 proteins to their sites and affect spliceosome activity, which results in the induction of AS (as illustrated in D). The authors supposed that lnc-RNA is transcribed in cells at a constant level, but the amount of EGPO and the grade of hTERT AS are regulated by EndoG activity and the fact that the translocation of EndoG into the cell nucleus triggers AS. Another result of studies on the identification of EGPO is that this oligonucleotide acts as a splice-switching oligonucleotide
[76], and the functioning of EndoG is the first observation of natural oligonucleotides that can modulate AS.
The described mechanism is not completely understood and requires further investigation. The most obvious issues are how lncRNA is transcribed from the noncoding strand of the hTERT gene; why EndoG creates an EGPO of this size as soon as other G-rich islands are present in lnc-RNA; why EGPO complements the junction site of exon 8 - intron 8 of hTERT pre-mRNA, and what other proteins are involved in this process. However, several facts favor the involvement of EndoG in this process. First, the suppression of EndoG expression by short interfering RNA abolished hTERT AS, and all hTERT presented was the full-length form
[72]. Second, EndoG and its translocation into the nucleus can be induced by different DNA-damaging agents. Cisplatin and other genotoxic agents demonstrated the ability to induce hTERT AS
[77]. Third, human cells display this mechanism, and the ability of EndoG to induce AS of TERT pre-mRNA was shown in rat and mouse cell lymphocytes
[78]. Fourth, transfection of cells with EGPO can induce hTERT AS
[62]. Fifth, EndoG is able to induce AS of the pre-mRNAs of other genes, the most studied of which are deoxyribonuclease 1 (DNase I)
[79], caspase-2 (Casp-2), and B-cell lymphoma X (BCL-x)
[80].