Selenium (Se) is a metalloid element that fulfills important physiological functions within the necessary dose, but human health is also vulnerable to selenium deficiency or selenium excess.
- diseases
- metabolism
- microRNA
- selenium
- selenoproteins
1. Introduction
Selenium (Se) is a metalloid element that fulfills important physiological functions within the necessary dose, but human health is also vulnerable to selenium deficiency or selenium excess [1]. Daily food can meet people’s demand for selenium, comprising a balanced selection of meat and plant products [2]. In the natural environment, rock and soil composition are believed to determine Se distribution characteristics [3]. Populations in the United States, Mexico, Colombia, India, and Iceland easily attain their recommended daily Se dose. However, populations in Northern Europe, Australia, New Zealand, and China have poor Se-containing soils, potentially leading to Se deficiencies [4].
Recently, microRNAs (miRNAs) and their role in Se-related inflammation and diseases have attracted considerable attention [5][6][7][8]. MiRNAs are non-coding endogenous single-stranded RNA molecules that consist of 20–23 nucleotides [9]. They play central roles in cell differentiation, proliferation, and survival by binding to complementary target messenger RNA (mRNA), leading to mRNA translation, inhibition, and/or degradation [10]. Therefore, they can be regarded as key gene expression regulators that can control physiological and pathological processes, including the development of cancer [9]. Studies have confirmed miRNA dysregulation is causal in many cancers [11][12][13][14][15], with miRNAs acting as tumor suppressors or oncogenes. Similarly, miRNA mimics and molecules targeting miRNAs have shown promise in preclinical studies [12].
Recently, microRNAs (miRNAs) and their role in Se-related inflammation and diseases have attracted considerable attention [5,6,7,8]. MiRNAs are non-coding endogenous single-stranded RNA molecules that consist of 20–23 nucleotides [9]. They play central roles in cell differentiation, proliferation, and survival by binding to complementary target messenger RNA (mRNA), leading to mRNA translation, inhibition, and/or degradation [10]. Therefore, they can be regarded as key gene expression regulators that can control physiological and pathological processes, including the development of cancer [9]. Studies have confirmed miRNA dysregulation is causal in many cancers [11,12,13,14,15], with miRNAs acting as tumor suppressors or oncogenes. Similarly, miRNA mimics and molecules targeting miRNAs have shown promise in preclinical studies [12].
Keshan disease (KD) and Kashin–Beck disease (KBD) are related to the Se deficiency, but the actual mechanisms that are behind these diseases are still not precisely understood [16]. Microarray and proteomics analysis revealed the genes and pathways that may be involved in these diseases. Nineteen Se- and three zinc-associated proteins were identified among 105 differentially-expressed proteins. The proteins involved in hypoxia-inducible factor-1α and apoptosis pathways may play significant roles in the pathogenesis of KD [17]. There are numerous functional pathways and cellular systems associated with the differentially expressed genes and proteins; the TCA Cycle II (Eukaryotic) pathway and NADP repair pathway may also participate in the pathogenesis of KD [18]. Thirty-four nutrients associated with differentially expressed genes and ten significant pathways have been identified for juvenile KBD, which are mainly related to metabolism, cell apoptosis, extracellular matrix, and other functions which consist of pathological changes of KBD [19]. One hundred and twenty-four miRNAs had lower expression levels in the subchondral bone sampled from KBD patients showed by miRNA array profiling [20]. These differential genes or proteins may become new targets for studying microRNAs in the two diseases.
2. Selenium Uptake and Metabolism
Se levels in a given food product does not mean an organism will derive its correct Se quota—instead, this depends on the bioavailability, bioaccessibility, and/or bioactivity of a given Se compound [2]. Human dietary Se forms mainly include organic and inorganic Se. These forms are typically absorbed without any regulatory processes and have a high bioavailability in the body [2][21]. Se is primarily absorbed in the duodenum and caecum after active transport via a sodium pump, but this process is different depending on the chemical form [22]. After intestinal absorption, different Se forms enter the bloodstream and are transported into the liver via the portal vein, where they are metabolized, transported, and distributed to different tissues [23] (
Se levels in a given food product does not mean an organism will derive its correct Se quota—instead, this depends on the bioavailability, bioaccessibility, and/or bioactivity of a given Se compound [2]. Human dietary Se forms mainly include organic and inorganic Se. These forms are typically absorbed without any regulatory processes and have a high bioavailability in the body [2,21]. Se is primarily absorbed in the duodenum and caecum after active transport via a sodium pump, but this process is different depending on the chemical form [22]. After intestinal absorption, different Se forms enter the bloodstream and are transported into the liver via the portal vein, where they are metabolized, transported, and distributed to different tissues [23] (Figure 1, adapted from [24][25][26][27] and drawn with
, adapted from [24,25,26,27] and drawn with access on 1 March 2021). In normal diets, tissue Se concentrations in the body range from the highest to the lowest in the following organs: Kidney, liver, spleen, pancreas, heart, brain, lung, bone, and skeletal muscle [28].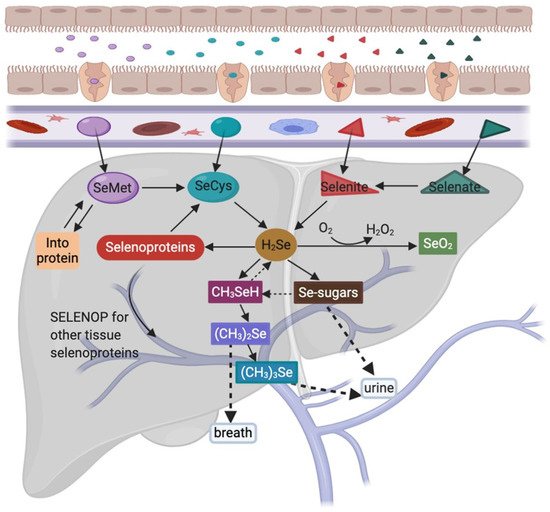
Figure 1.
2
3
3
2
3
3
+
2
SeMet accounts for 90% of total Se in plants. Some SeMet is randomly incorporated into proteins at methionine positions [29], whereas other SeMet quantities are metabolized to selenocysteine (SeCys) via methionine cycle and transsulfuration pathways in the liver [26]. Furthermore, Se-methylselenocysteine and γ-glutamyl-Se-methylselenocysteine (believed to exert anticancer effects) are also found in plants, such as garlic, onions, and broccoli, and they are metabolized to methyl selenol [25][30]. SeCys occurs at much lower levels than SeMet in plants. When SeCys is absorbed, free SeCys does not appear to generate concentrations for efficient attachment to cysteine transfer RNA (tRNA). But once incorporated into proteins, SeCys predisposes these proteins to degradation processes [31]. SeCys is the main Se source in animal products, however, highly reactive free SeCys is maintained at very low concentrations in tissues [32]. Inorganic Se is the main form of Se supplementation as it promotes selenoprotein biosynthesis [26]. Selenate must be reduced to selenite before further metabolism. Then, interactions with the tripeptide and glutathione ensure this selenite is reduced to selenide (H
SeMet accounts for 90% of total Se in plants. Some SeMet is randomly incorporated into proteins at methionine positions [29], whereas other SeMet quantities are metabolized to selenocysteine (SeCys) via methionine cycle and transsulfuration pathways in the liver [26]. Furthermore, Se-methylselenocysteine and γ-glutamyl-Se-methylselenocysteine (believed to exert anticancer effects) are also found in plants, such as garlic, onions, and broccoli, and they are metabolized to methyl selenol [25,30]. SeCys occurs at much lower levels than SeMet in plants. When SeCys is absorbed, free SeCys does not appear to generate concentrations for efficient attachment to cysteine transfer RNA (tRNA). But once incorporated into proteins, SeCys predisposes these proteins to degradation processes [31]. SeCys is the main Se source in animal products, however, highly reactive free SeCys is maintained at very low concentrations in tissues [32]. Inorganic Se is the main form of Se supplementation as it promotes selenoprotein biosynthesis [26]. Selenate must be reduced to selenite before further metabolism. Then, interactions with the tripeptide and glutathione ensure this selenite is reduced to selenide (H2
Se), which is a central gateway molecule for Se utilization and excretion [25]. Furthermore, all seleno-compounds must be metabolized to selenide for incorporation into selenoproteins [29]. After initial Se metabolism, H2
Se is converted to selenophosphate, which is used to convert phosphoseryl-tRNA[Ser]Sec
to SeCys-tRNA[Ser]Sec
. Then the SeCys-tRNA[Ser]Sec
reads the UGA codon and integrates SeCys into the amino acid sequence to form a selenoprotein [33]. Selenoprotein P, which is mainly produced in the liver, transports Se from the liver to extrahepatic tissues and organs, where it is metabolized to prevent oxidative damage [26]. Methyl selenol is demethylated to H2
Se in the equilibrium reaction, where it and its precursors (SeMet and CH3
SeCys) may be used as Se sources for selenoprotein synthesis [34]. The oxidation of excess H2
Se leads to superoxides and other active oxygen species, often with toxic effects [35]. Se excess detoxification occurs via sequential methylation into dimethyl selenide, and is excreted via the breath, whereas Se-sugars and trimethyl selenonium are excreted in the urine [27]. Although all Se forms are excreted from the body at some stage, only Se-sugars are bioavailable. It is not only an excretion metabolite of Se, but also may transport selenium from liver cells to other cells in the body [36].3. Selenium Related Pathogenic Mechanisms and Diseases
In general, when Se plasma levels are less than 85 µg/L, Se deficiency becomes evident in the body [37]. Se deficiency is caused by poor Se dietary intake, and may be induced or aggravated by nutritional, chemical, and infectious stresses. Several Se deficiency animal diseases are related to the co-existing vitamin E deficiencies [38]. Se deficiency causes heart disease (e.g., cardiomyopathy, arrhythmias), infertility, neuronal or neuromuscular diseases, and increased susceptibility to cancer, infection, and heavy metal toxicity [39][40][41][42][43]. The maximum harmless Se concentration is less than 400 μg per day in adults [44]. Excess dietary Se causes adverse effects (selenosis), including acute food poisoning symptoms, such as vomiting, nausea, and diarrhea, as well as chronic toxicity manifested by the hair and nail brittleness and loss, gastrointestinal disturbances, infertility, and nervous system abnormalities [45]. In addition, when Se is excessive, the toxicity of inorganic Se is much lower than that of organic selenomethionine (SeMet) [38].
In general, when Se plasma levels are less than 85 µg/L, Se deficiency becomes evident in the body [37]. Se deficiency is caused by poor Se dietary intake, and may be induced or aggravated by nutritional, chemical, and infectious stresses. Several Se deficiency animal diseases are related to the co-existing vitamin E deficiencies [38]. Se deficiency causes heart disease (e.g., cardiomyopathy, arrhythmias), infertility, neuronal or neuromuscular diseases, and increased susceptibility to cancer, infection, and heavy metal toxicity [39,40,41,42,43]. The maximum harmless Se concentration is less than 400 μg per day in adults [44]. Excess dietary Se causes adverse effects (selenosis), including acute food poisoning symptoms, such as vomiting, nausea, and diarrhea, as well as chronic toxicity manifested by the hair and nail brittleness and loss, gastrointestinal disturbances, infertility, and nervous system abnormalities [45]. In addition, when Se is excessive, the toxicity of inorganic Se is much lower than that of organic selenomethionine (SeMet) [38].
Both Se excess and deficiency lead to Se-related disease. A recent epidemiological analysis showed that taking 300 µg/d selenium for 5 consecutive years increased all-cause mortality after 10 years in countries with moderately low selenium levels [46]. Dietary Se functions mainly depend on the selenoprotein form that exerts biological effects in the body. Almost all tissues are affected by changes in Se status or selenoprotein expression [47][48][49]. For instance, embryonic lethality caused by trsp gene deletion, which encodes Sec-tRNA for translation, also reflects the importance of selenoproteins to the body [50]. Currently, 25 genes in the human genome have been identified as encoding selenoproteins, with most exhibiting antioxidant activities [51]. Other specific processes include the biosynthesis of deoxyribonucleoside triphosphates for DNA, the reduction of oxidized proteins and membranes, redox regulation of transcription factors, apoptosis regulation, immunomodulation, thyroid hormone regulation, Se transport and storage, protein folding, and the degradation of misfolded proteins in the endoplasmic reticulum [27]. Therefore, it appears the physiological and pathological changes or diseases caused by Se deficiency are primarily mediated by a selenoprotein imbalance [28][52][53][54][55]. Correspondingly, excess Se generates toxicity via several mechanisms [56]: (1) CH
Both Se excess and deficiency lead to Se-related disease. A recent epidemiological analysis showed that taking 300 µg/d selenium for 5 consecutive years increased all-cause mortality after 10 years in countries with moderately low selenium levels [46]. Dietary Se functions mainly depend on the selenoprotein form that exerts biological effects in the body. Almost all tissues are affected by changes in Se status or selenoprotein expression [47,48,49]. For instance, embryonic lethality caused by trsp gene deletion, which encodes Sec-tRNA for translation, also reflects the importance of selenoproteins to the body [50]. Currently, 25 genes in the human genome have been identified as encoding selenoproteins, with most exhibiting antioxidant activities [51]. Other specific processes include the biosynthesis of deoxyribonucleoside triphosphates for DNA, the reduction of oxidized proteins and membranes, redox regulation of transcription factors, apoptosis regulation, immunomodulation, thyroid hormone regulation, Se transport and storage, protein folding, and the degradation of misfolded proteins in the endoplasmic reticulum [27]. Therefore, it appears the physiological and pathological changes or diseases caused by Se deficiency are primarily mediated by a selenoprotein imbalance [28,52,53,54,55]. Correspondingly, excess Se generates toxicity via several mechanisms [56]: (1) CH
3Se− formation, which either enters a redox cycle and generates superoxide and oxidative stress, or generates free radicals that bind to and inhibit key enzymes and proteins, (2) SeCys excess, which inhibits Se methylation metabolism, results in hydrogen selenide (intermediate metabolite) accumulation eventually leading to hepatotoxicity and other Se-related adverse effects, (3) excess Se analogs of sulfur-containing enzymes and structural proteins also play roles in avian teratogenesis. Equally, aquatic organisms exposed to high Se doses are at risk of organ damage and genomic mutations, which potentially pose a threat to human food chains [57][58].
Se− formation, which either enters a redox cycle and generates superoxide and oxidative stress, or generates free radicals that bind to and inhibit key enzymes and proteins, (2) SeCys excess, which inhibits Se methylation metabolism, results in hydrogen selenide (intermediate metabolite) accumulation eventually leading to hepatotoxicity and other Se-related adverse effects, (3) excess Se analogs of sulfur-containing enzymes and structural proteins also play roles in avian teratogenesis. Equally, aquatic organisms exposed to high Se doses are at risk of organ damage and genomic mutations, which potentially pose a threat to human food chains [57,58].
4. Current Progress in Nutrient Regulation of miRNAs
4.1. miRNAs
MiRNAs are derived from intergenic or intragenic (exon and intron) genomic regions [59]. They are usually transcribed by RNA polymerase II from miRNA genes, first forming a ‘primary miRNA transcript’ (pri-miRNA). This transcript is cleaved by a microprocessor complex, comprising the double-stranded RNase III enzyme, DROSHA, and its essential cofactor, the DiGeorge syndrome critical region 8 (DGCR8) protein, generating a short sequence, the ‘miRNA precursor’ (pre-miRNA), which displays a hairpin-like secondary structure [60]. The pre-miRNA is exported to the cytoplasm and processed by DICER, a ribonuclease III enzyme that produces the mature miRNA for final incorporation into an RNA-induced silencing complex (RISC) [61]. Under most conditions, mature RISC represses gene expression post-transcriptionally by binding to 3′ untranslated regions of specific mRNAs, and mediates degradation, destabilization, or translational inhibition, based on target sequence complementarity [62]. MiRNAs are abundant in all cells, are found in extracellular body fluids (e.g., serum, plasma, saliva, and urine), and are implicated in several pathological conditions [63][64][63,64]. During some biological processes, miRNAs regulate protein levels of key regulatory factors, or serve as switches to govern gene expression [65]. Post-transcriptional regulation of miRNA can improve the compliance, accuracy, and sensitivity of gene expression regulation [66]. Due to its small molecular size, each miRNA potentially targets hundreds of mRNA molecules [67]. Similarly, each mRNA may be targeted by multiple miRNAs to form complexes and multifaceted regulatory networks [68]. Moreover, miRNAs also regulate DNA methylation and histone modification [69]. It was previously reported that >60% of human coding genes are regulated by miRNAs [70], and >2800 mature miRNA sequences are described in the miRBase 22 repository (http://www.mirbase.org/ access on 5 February 2021). In addition, miRNAs are not only endogenously synthesized, but may be derived from the diet (e.g., milk and plants) [71].
4.2. Regulation Mechanisms of Vitamins and Minerals on miRNAs
4.3. Role of Mammalian Target of Rapamycin (mTOR) in Nutrient Regulation of miRNAs
4.4. miRNAs Mediated by Se Status Are Implicated in Disease Development and Progression
In this section, recent developments on miRNA involvement in Se-related diseases are described, including miRNA changes when Se is in deficient, moderate, or excessive status, regulatory miRNA targets, and miRNA roles in Se antioxidant damage or Se-related diseases (
).
Se supplementation reportedly changed miRNA profiles in the intestinal cell line, Caco-2, differentially down-regulating 12 miRNAs under nutrient deficiency conditions [77]. The miRNAs most affected were miR-185, miR-625, miR-203, and miR-429, whereas pathway analyses identified arachidonic acid metabolism, glutathione metabolism, oxidative stress, and mitochondrial respiration as Se-sensitive pathways [88]. In a Se deficiency rat model, five miRNAs from harvested heart tissue (miR-374, miR-16, miR-199a-5p, miR-195 and miR-30e were up-regulated > 5-fold in the Se nutrient deficiency group, when compared with the Se-supplemented group, whereas three miRNAs were down-regulated (miR-3571, miR-675, and miR-450a [78]. Up-regulated miRNAs were involved in signal transduction, cell differentiation, and stress responses, suggesting roles in cardiac function and regulation [89]. A Se pro-longevity mechanism study reported that several miRNAs were altered in response to dietary Se in the mouse liver [3]. Expression levels of 38 miRNAs were altered by Se deficiency compared with Se sufficiency, and the study showed that selenoprotein regulation by miRNAs was not a direct effect [3]. The role of glutathione peroxidase regulation and related miRNAs has also been reported [90][91]. In an intervention study in elderly males given Se and coenzyme Q10 supplements for four years, significant expression differences were observed in >100 miRNAs, with up to 4-fold differences in combined Se and coenzyme Q10 supplementation experiments [92]. Such changes may contribute to underlying clinical mechanisms. Early reports indicated that cardiovascular mortality was reduced, cardiac function improved, and inflammation and oxidative stress indications decreased after Se intervention [92]. Se decreased inflammation by increasing miR-146a expression, decreasing mmu-miR-155, TLR2/6, NF-κB, and MAPK signaling pathway expression in mammary tissue from infected animals, and mammary epithelial cells [93][94]. Although these studies investigated miRNA-mediated Se deficiency or Se antioxidant damage, data for miRNAs implicated in Se excess are limited. When Se is in excess, it potentially increases the risk of metabolic syndrome [95][96].
Se supplementation reportedly changed miRNA profiles in the intestinal cell line, Caco-2, differentially down-regulating 12 miRNAs under nutrient deficiency conditions [77]. The miRNAs most affected were miR-185, miR-625, miR-203, and miR-429, whereas pathway analyses identified arachidonic acid metabolism, glutathione metabolism, oxidative stress, and mitochondrial respiration as Se-sensitive pathways [88]. In a Se deficiency rat model, five miRNAs from harvested heart tissue (miR-374, miR-16, miR-199a-5p, miR-195 and miR-30e were up-regulated > 5-fold in the Se nutrient deficiency group, when compared with the Se-supplemented group, whereas three miRNAs were down-regulated (miR-3571, miR-675, and miR-450a [78]. Up-regulated miRNAs were involved in signal transduction, cell differentiation, and stress responses, suggesting roles in cardiac function and regulation [89]. A Se pro-longevity mechanism study reported that several miRNAs were altered in response to dietary Se in the mouse liver [3]. Expression levels of 38 miRNAs were altered by Se deficiency compared with Se sufficiency, and the study showed that selenoprotein regulation by miRNAs was not a direct effect [3]. The role of glutathione peroxidase regulation and related miRNAs has also been reported [90,91]. In an intervention study in elderly males given Se and coenzyme Q10 supplements for four years, significant expression differences were observed in >100 miRNAs, with up to 4-fold differences in combined Se and coenzyme Q10 supplementation experiments [92]. Such changes may contribute to underlying clinical mechanisms. Early reports indicated that cardiovascular mortality was reduced, cardiac function improved, and inflammation and oxidative stress indications decreased after Se intervention [92]. Se decreased inflammation by increasing miR-146a expression, decreasing mmu-miR-155, TLR2/6, NF-κB, and MAPK signaling pathway expression in mammary tissue from infected animals, and mammary epithelial cells [93,94]. Although these studies investigated miRNA-mediated Se deficiency or Se antioxidant damage, data for miRNAs implicated in Se excess are limited. When Se is in excess, it potentially increases the risk of metabolic syndrome [95,96].
Apart from the aforementioned transcription factors being implicated in miRNA regulation by Se, it remains unclear how Se precisely regulates miRNAs. Thus, similar to mechanisms involved in nutrition-gene interactions, it is reasonable to speculate that selenium regulates the expression of miRNAs by potentially affecting the epigenetic regulation mechanisms, including DNA methylation and histone modification. Se supplementation may modify global DNA methylation and specific gene regions, possibly via DNMT inhibition [97]. Additionally, dietary Se deficiency may decrease DNA methylation by enhancing trans-sulfonation pathways [98]. Se also alters histone modification via HDAC inhibition of the Se metabolism products, seleno-α-keto acids [97]. Taken together, the current evidence indicates that different DNA hypomethylation mechanisms occur at different Se levels, including (1) the redirection of homocysteine towards transsulfuration pathways and glutathione synthesis during Se deficiency, (2) excess Se competes with S-adenosylmethionine to use the methyl group required for selenium metabolism—consequently, S-adenosylmethionine levels are reduced for DNMT and methylation processes are similarly inhibited and (3) Se affects specific tumor suppressor gene methylation mechanisms, possibly in a sex-dependent manner. Importantly cancer phenotypes are often characterized by the altered methylation of selenoprotein-encoding genes, mainly glutathione peroxidase 3 [99].
Table 1.
MiRNAs are regulated by selenium (deficiency, moderate, and excess).
| Se Status | Micro RNA | Target | Observed Effect | Note | Reference | ||
|---|---|---|---|---|---|---|---|
| Se deficiency | ↑miR-181a-5P | ↓SBP2 | ↓GPX1, GPX4, and SELENOS levels | In C28/I2 human juvenile chondrocytes and DA rats | [100] | ||
| ↑Gga-let-7f-3p | ↓SELENOK | ↑Oxidative stress, ERS, and apoptosis | In chicken myoblasts and muscle | [101] | |||
| ↑miR-200a-5p | ↓ | TXNRD2, TXNRD3, SELENON, SELENOT, SELENOF | and | SELENOP | ↑Glucose metabolism disorder, cardiomyocyte hypertrophy | In chicken cardiomyocytes | [102] |
| ↓RNF11 | ↑Oxidative stress and myocardial necroptosis | In chicken cardiac tissue and cardiomyocytes | [103] | ||||
| ↑miR-138-5p | ↓SELENOM | ↑Apoptosis, oxidative stress, mitochondrial fission | In chicken chondrocytes | [104] | |||
| ↑miR-544a | ↓SELENOK | Interferes with SELENOK translation | In HepG2 and HuH-7 human hepatocarcinoma cells | [105] | |||
| ↑miR-196-5p | ↓NFκBIA (IκB-α) | ↑LPS-induced oxidative stress and inflammation, respiratory mucosal immune dysfunction | In chicken trachea | [106] | |||
| ↑miR-193b-3p | ↓MAML1 | ↑Hepatocyte apoptosis | In the liver tissues and primary hepatocytes from broilers | [107] | |||
| ↑miR-33-3p | ↓ADAM10 | ↑Cell cycle arrest and apoptosis | In vivo and in vitro in the chicken kidney | [108] | |||
| ↓E4F1 | ↑Oxidative stress, ERS, and apoptosis | In vein endothelial cells from broilers | [109] | ||||
| ↑miR-328 | ↓ATP2A2 | ↑Intracellular Ca | 2+ | and cell apoptosis | In H9c2 rat cardiac myoblasts | [110] | |
| ↑miR-215-5p | ↓CTCF | ↑Mitochondrial biosynthesis imbalance, defects in myocardial development | In heart tissue and primary cardiomyocytes from chickens | [111] | |||
| ↓PI3K/AKT/TOR | ↑ROS, Myocardial autophagy | In cardiomyocytes of chicken | [112] | ||||
| ↑miR-1594 | ↓TNNT2 | ↑Ca | 2+ | In heart and primary cardiomyocytes from chickens | [113] | ||
| ↑miR-2954 | ↓PI3K | ↑Autophagy and apoptosis | In heart and primary cardiomyocytes from chickens | [114] | |||
| ↑miR-16-5p | ↓PI3K/AKT | ↑Necroptosis | In tracheal tissues and tracheal epithelial cells of chicken | [115] | |||
| ↑miR-128-1-5p | ↓CADM1 | ↑Tight junction structural damage and cell cycle arrested | In vein tissues and vein endothelial cells from broilers | [116] | |||
| ↑ miR-374, miR-16, miR-199a-5p, miR-195 and miR-30e | ↓ miR-3571, miR-675a and miR-450a | ↑ Wnt/β--catenin | ↑ Cardiac dysfunction | In rat heart | [89] | ||
| ↓miR--185 | ↑GPX2, SEPHS2 | ↑Altered expression of 12 miRNA and 50 genes | In Caco-2 human intestinal cells | [88] | |||
| ↓miR-29a-3p | ↑TNFR1 | Altered expression of selenoprotein genes, ↑necrotic cells | In the pig brain and IPEC-J2 pig intestinal epithelial cells | [117] | |||
| ↓miR-155 | ↑TNFRSF1B | ↑Oxidative stress-induced apoptosis | In splenic cells and spleen of broilers | [118] | |||
| ↓miR-146a | ↑MAPKs | ↑ROS-induced inflammation | In the head kidney of carp | [119] | |||
| ↓miR-7 | ↓SELENOP | Both are potential biomarkers of HCC | In HCC patients and HepG2 human hepatocarcinoma cells | [120] | |||
| Se moderate | ↑miR-146a | ↓TLR2, TLR6, NF-κB and MAPK | ↓ | S. aureus | -infected mastitis | In mammary tissues and mammary epithelial cells from mouse | [93] |
| ↑miR-125a and miR-125b | ↓Bak and caspase-3 | ↓Cd-induced apoptosis | In LLC-PK1 porcine renal epithelial cells | [121] | |||
| ↑ miR-29b-3p, miR-30e-5p and miR-19a-3p | ↓ miR-199a-5p, miR-130a-3p and miR-191-5p | —— | ↓ Risk of heart failure | In healthy elderly males | [92] | ||
| ↓mmu-miR-155 | ↓TNF-α, IL-1β, IL-10, TLR2, NF-κB and MAPKs | ↓ | S. aureus | -infected mastitis | In mammary tissues and mammary epithelial cells from mouse | [94] | |
| ↓miR-224 | ↑ | ID1 | ↓Pb-induced oxidative damage and restoring thyroid hormone disequilibrium | In thyroid tissues of male rats | [122] | ||
| ↓miR-16-5p | ↑PiK3R1 and IGF1R | ↓Pb-induced neutrophil apoptosis | From chicken peripheral blood | [123] | |||
| ↓miR-216a | ↑PI3K/AKT | ↓Cd-triggered necrosis and apoptosis | In the splenic lymphocytes of common carp | [124] | |||
| Se excess | ↑miR-122-5p | ↑BMI, SBP and DBP | ↑Risk of MetS | In male adults | [95] | ||
| ↑miR-454-3p and miR-584-5p | ↓miR-375 | A link between Se intake, vitamin D metabolism, and calcium homeostasis | ↑miR-375 as a potential biomarker of MetS | In obese women | [96] |
miRNAs, genes, or proteins down-regulated/inhibited (↓) or up-regulated/activated (↑). SBP2, SECIS binding protein 2; SELENOK, selenoprotein K; TXNRD2, thioredoxin reductase 2; TXNRD3, thioredoxin reductase 3; SELENON, selenoprotein N; SELENOT, selenoprotein T; SELENOF, selenoprotein F; SELENOP, selenoprotein P; RNF 11, ring finger protein 11; SELENOM, selenoprotein M; NFκBIA (IκB-α), IkappaB-alpha; MAML1, mastermind-like protein 1; ADAM 10, adisintegrin and metalloprotease domain 10; E4F1, E4F transcription factor 1; ATP2A2, sarcoplasmic/endoplasmic reticulum calcium ATPase 2; CTCF, CCCTC-binding factor; TNNT2, Troponin T Type 2; CADM1, cell adhesion molecule 1; GPX2, glutathione peroxidase 2; SEPHS2, selenophosphatesynthase 2; TNFR1, TNF receptor superfamily member 1A; TNFRSF1B, TNF receptor superfamily member 1B; TLR2, toll-like receptor 2; TLR6, toll-like receptor 6; ID1, Inhibitor of DNA binding 1; PIK3R1, phosphoinositide-3-kinase regulatory subunit 1; IGF1R, type 1 insulin-like growth factor receptor; BMI, body mass index; SBP, systolic pressure, and DBP, diastolic pressure.