Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Anton Gorkovskiy and Version 2 by Catherine Yang.
Mutations in DNA can be limited to one or a few nucleotides, or encompass larger deletions, insertions, duplications, inversions and translocations that span long stretches of DNA or even full chromosomes. These so-called structural variations (SVs) can alter the gene copy number, modify open reading frames, change regulatory sequences or chromatin structure and thus result in major phenotypic changes.
- structural variation
- fungi
- adaptation
1. Introduction
Structural variation (SV) groups different forms of mutations that involve longer stretches of DNA, including deletions, insertions, duplications, inversions, translocations, or even full chromosome fusion, fission or loss (Figure 1). Structural variants can be balanced and show no specific loss or gain of DNA information, such as inversions of a genetic fragment or translocations of a stretch of DNA within or between chromosomes, or they can be unbalanced, where a part of the genome is lost (deletions), acquired (insertions) or duplicated (duplications), which is termed copy number variation (CNV).
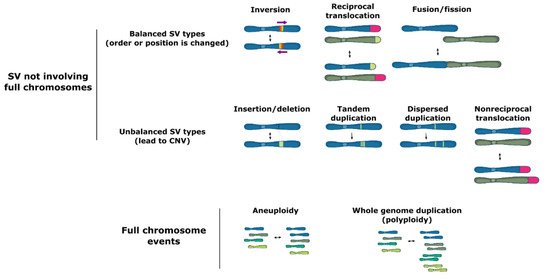
Figure 1. Types of structural variation.
Structural variation may occur both in coding and noncoding regions of the genome, including in highly repetitive elements, such as transposons. SV events can lead to major phenotypic changes via diverse mechanisms including modification of open reading frames, changes in gene expression due to copy number variation, alteration of regulatory sequences (via gain or loss of functional genomic elements) or chromatin structure, or even formation of novel genes [1][2][3][4][5][1,2,3,4,5]. Moreover, some forms of SV, such as large inversions and chromosomal fusions, cause a reduction in recombination rates between homologous chromosome pairs. In turn, the reduced recombination may facilitate the cosegregation of multiple adaptive polymorphisms as if they were controlled by a single genetic locus (linkage disequilibrium and supergene formation) [6][7][8][9][10][11][6,7,8,9,10,11].
In humans, single nucleotide variants (SNVs) are the most common type of variation, but SV accounts for a higher number of variable nucleotides between genomes, with roughly 0.5% of the human genome being involved in structural variation [12][13][12,13]. Strikingly, third-generation (long-read) genome sequencing of a clonal population of seven closely related Schizosaccharomyces pombe strains that diverged ∼50–65 years ago revealed that they have an average pairwise difference of 19 SNVs and four nonoverlapping larger duplications [14]. Moreover, SVs are three times more likely to be associated with a genome-wide association signal and 50 times more likely to be associated with expressed quantitative trait loci than single nucleotide variants, further hinting at their importance as drivers of phenotypic variation [13][15][13,15]. Importantly, despite the significant contribution of SV events (especially of CNVs) to quantitative traits, they are frequently overlooked in studies employing short-read sequencing technologies [14].
The phenotypic consequences of SVs have traditionally been assumed to be almost exclusively negative. This is perhaps partly due to the association of SVs with many human diseases, especially autoimmune, metabolic, and cognitive disorders [16][17][18][19][20][16,17,18,19,20]. However, the emergence of advanced genotype-to-phenotype mapping technologies, as well as studies focusing on experimental evolution have led to a growing body of evidence suggesting that many SVs are neutral or even adaptive, both in humans [12][15][21][12,15,21] and other organisms, including microbes [11][22][23][24][25][26][27][28][29][30][31][11,22,23,24,25,26,27,28,29,30,31]. SVs are therefore increasingly considered to be an important evolutionary driver, and some studies suggest that SV may be especially important for quick adaptation.
2. Mechanisms of SV Formation
SV involving complete chromosomes is often caused by defective chromosome segregation. Chromosomes must be meticulously replicated and equally segregated at each cell division. Distortion of either one of these processes can lead to SV formation. In particular, failure of any of the critical chromosome segregation steps, including chromatid cohesion, spindle pole body (functional equivalent of the mammalian centrosome) formation at opposite cell poles, kinetochore–microtubule attachment, and quality control at the spindle assembly checkpoint can result in aneuploidy (i.e., loss or gain of whole chromosomes) (Figure 2A) [32].
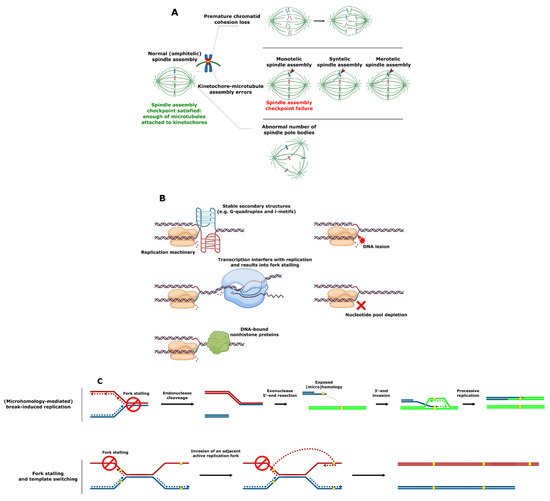
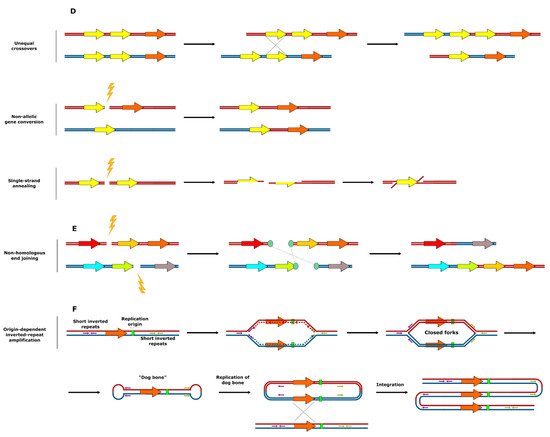
Figure 2. Mechanisms of SV formation. (A) Events leading to aneuploidy. (B) Events leading to replication fork collapse. (C) SV formation as a result of stalled replication fork reactivation. (D) SV formation mediated by homologous recombination. (E) SV formation mediated by nonhomologous end joining. (F) Origin-dependent inverted-repeat amplification.
An SV that does not involve full chromosomes often results from compromised DNA replication, where processive forks collide with the replication fork barriers (Figure 2B) [33][34][35][33,34,35]. These barriers typically include (1) specific DNA secondary structures such as G-quadruplex (G4) motifs [36][37][38][36,37,38], which are enriched in the telomeres, ribosomal DNA (rDNA) and promoter regions in S. cerevisiae, Schizosaccharomyces pombe, and human cells [39][40][41][42][43][44][39,40,41,42,43,44]; (2) highly expressed loci such as the tRNA genes where transcription can interfere with replication [45][46][47][45,46,47]; or (3) tightly DNA bound nonhistone proteins (e.g., at centromeres) [48][49][48,49]. Replication forks can also be stalled as a result of DNA damage or the inhibition of replication by nucleotide depletion [50][51][50,51]. Reactivation of blocked replication forks and DNA damage can lead to SV due to the occurrence of nonallelic homologous recombination resulting from incorrect repair template utilization (Figure 2C) [52][53][54][52,53,54]. This process is remarkably more frequent in the case of dispersed repetitive DNA sequences such as transposable elements or remnants of those (long terminal repeats), tRNA genes, origins of replication, and clusters of tandemly repeated genes including those encoding ribosomal RNA and those residing in subtelomeric duplication blocks [14][54][55][56][57][58][59][60][61][62][63][64][65][66][67][14,54,55,56,57,58,59,60,61,62,63,64,65,66,67]. Curiously, stretches of repetitive DNA and, in particular, transposable elements are enriched in highly fast-evolving genomic compartments (which exist as ‘islands’ on core chromosomes) and accessory chromosomes of many pathogenic filamentous fungi [68][69][70][71][72][73][74][68,69,70,71,72,73,74]. These genomic compartments were shown to be the hot spots of SV [73][74][73,74]. Increased plasticity of the indicated genomic regions known to bear the virulence-related genes likely allows pathogens to keep up with the evolution of the host defense mechanisms and succeed in pathogen–host “arms race”.
A third major mechanism underlying SV is linked to crossing-over between repetitive DNA sequences and the repair of DNA double strand breaks near repetitive DNA sequences (Figure 2D,E) [75][76][75,76]. Various types of homologous recombination at repeat sites, including unequal crossovers, gene conversion, and single-strand annealing were reported to result in CNV [75]. A specific example of repeat-associated CNV generation, origin-dependent inverted-repeat amplification (Figure 2F), was hypothesized to underlie the amplification of the SUL1 locus in yeast [77][78][77,78]. As a result of the DNA replication error at small, interrupted inverted repeats, nascent leading and lagging strands get covalently linked. This ends up in formation of an extrachromosomal circular intermediate, and its integration into original chromosomal locus results in the gene triplication [77][78][77,78]. In some specific cases, copy number amplification is achieved via the formation of the extrachromosomal circular elements [79], which were proposed to be a fast and revocable mechanism of gene copy number amplification [80][81][80,81].
Finally, a very specific source of gene duplications is the whole genome duplication (WGD, also referred to as polyploidization)—i.e., addition of a complete set of chromosomes to the genome [82][83][82,83].
3. Balanced SV Events
Balanced SV types such as reciprocal translocations and inversions are widespread in Saccharomyces species and other fungi [14][60][63][65][71][72][84][85][86][87][88][89][14,60,63,65,71,72,224,225,226,227,228,229]. They are thought to serve as initial genetic barriers in eukaryotic speciation and, thus, to contribute to the onset of reproductive isolation and speciation [90][91][92][93][94][95][96][230,231,232,233,234,235,236]. In flies [97][237], mosquitoes [98][238], and flowering plants [8], inversions are hypothesized to also play a role in evolutionary adaptation. Analysis of the outcomes of chromosomal translocations in S. cerevisiae [99][239] and of translocations and inversions in S. pombe [85][100][225,240] demonstrated that these types of SV can significantly influence the fitness of the organism in specific environments, possibly as some events cause changes in gene expression [85][225]. It was hypothesized that balanced types of SV can be maintained as polymorphisms in nature despite their meiotic costs (low viability in heterozygotic crosses) when this disadvantage is outweighed by the fitness advantage gained in mitosis (antagonistic pleiotropy) [85][225]. Contrastingly, Naseeb and colleagues were not able to detect phenotypic consequences of a set of large inversions, even if they did observe significant changes in gene transcription patterns [101][241]. This again underscores that the effect of a specific structural rearrangement always depends on the affected genetic locus, the genetic background and the environment.