Essential hypertension (EH) is a highly heterogenous disease with a complex etiology. Toll-like receptors (TLRs) have been implied as novel effectors in this inflammatory environment since they can significantly stimulate the production of pro-inflammatory cytokines, the migration and proliferation of smooth muscle cells and the generation of reactive oxygen species (ROS), facilitating a low-intensity inflammatory background that is evident from the very early stages of hypertension. Furthermore, the net result of their activation is oxidative stress, endothelial dysfunction, vascular remodeling, and finally, vascular target organ damage, which forms the pathogenetic basis of EH. Importantly, evidence of augmented TLR expression and activation in hypertension has been documented not only in immune but also in several non-immune cells located in the central nervous system, the kidneys, and the vasculature which form the pathogenetic core systems operating in hypertensive disease.
- hypertension
- innate immunity
- toll-like receptors
- inflammation
- cytokines
1. Introduction
Essential hypertension (EH) has an independent and continuous relationship with the incidence of several cardiovascular events, and it is a leading cause of cardiovascular morbidity and mortality, surpassing all other cardiovascular risk factors such as diabetes mellitus, smoking, and hyperlipidemia [1][2]. In particular, the global burden of EH currently affects almost 30 to 45% of all adults and accounts for almost 10 million deaths and over 200 million disability-adjusted life years worldwide, with a constantly increasing tendency [3][4]. The continuous and close relationship of hypertension with cardiovascular disease, along with its overall prevalence worldwide, clearly render EH a major issue of public health that calls for immediate action.
Despite advances in awareness and pharmacotherapy, no unifying mechanism, and thus, no single therapeutic target exists, rendering control rates in the general hypertensive population unacceptably low. This is due to the fact that EH is a highly heterogenous disease with a multifactorial and complex etiology, still not fully elucidated [5]. Recently, chronic inflammation triggered and sustained by excessive immune system activation has been implicated as a novel contributor to the pathogenesis of the disease [6]. Towards this direction, accumulating evidence has highlighted the role of toll-like receptors (TLRs) as major effectors of innate immunity, in promoting inflammation, oxidative stress, and endothelial dysfunction—all major pathophysiologic components mediating vascular damage in EH [7][8][9][10].
2. Pathogenesis of Hypertension
It has been strongly supported that the pathogenesis of EH mainly consists of a noxious interplay between sophisticated neural, vascular, renal, and hormonal mechanisms, of which increased activation of the sympathetic nervous (SNS) and the renin-angiotensin system (RAS) prevail [5]. However, in recent years, it has been advocated that the core pathophysiologic event behind most of these mechanisms operating at the biological level is inflammation [11]. More specifically, a growing amount of evidence has demonstrated that multiple inflammatory mechanisms including pro-inflammatory cytokine and chemokine expression, cell infiltration and oxidative stress—all triggered and sustained by excessive immune system activation—are highly upregulated in the hypertensive environment [6][12].
3. The Role of the Immune System in the Pathogenesis of Hypertension
The immune system consists of both innate and adaptive immunity. Innate immunity represents the first line of defense of the human body and its main effector cells include dendritic cells, macrophages, granulocytes, natural killer (NK) cells, B cells, and mast cells, all of which act rapidly and non-specifically once activated by the presenting antigens. In contrast, adaptive immunity consists of T and B lymphocytes that mainly depend upon antigen recognition by the antigen-presenting cells in order to elicit a more robust inflammatory response [13].
Both systems are actively implicated in the pathogenesis of EH. More specifically, it has been hypothesized that hypertensive stimuli including angiotensin II (AngII), aldosterone, endothelin-1, salt, and several genes (i.e., ADRA2A [10q24-q26] (which is the predominant subtype gene modulating SNS outflow in the brain), ADRA2C [4p16.1], and ADRA2B [2p13-q13]) [14][15] increase central nervous signaling and thus SNS activity, which, in turn, provokes slight elevations in blood pressure (BP) [11]. This initial event leads to tissue injury and the release of endogenous intra- or extracellular molecules including cell-derived nucleic acids, fatty acids, heat shock proteins (HSPs), and high-mobility group box-1 (HMGB1), all termed damage-associated molecular patterns (DAMPs). Under normal conditions, DAMPs represent an acute “alarm signal”, warning the host to activate its defense mechanisms of which innate immunity is the first and most crucial one. In the hypertensive environment, though, it has been hypothesized that the pronounced DAMP-mediated stimulation of the immune system leads to significant inflammatory responses through two distinct pathways: i) the direct stimulation of innate immune cells further activating deleterious mechanisms including organ infiltration, chemokine and pro-inflammatory cytokine production, oxidative stress, phagocytosis, and complement activation, or ii) the consequent activation of adaptive immunity [6]. Activation of the adaptive immune response leads to the polarization of naive CD4+ T helper lymphocytes (Th0) towards pro-inflammatory T helper Th1 and Th17 phenotypes that produce reactive oxygen species (ROS), interferon (IFN)-γ, and interleukin (IL)-17 [16]. The net effect is a state of chronic low-grade inflammation that leads to additional vascular dysfunction, increased BP, tissue injury, and finally, the release of more DAMPs, which, in turn, flare up and maintain immune system hyperresponsiveness [17]. As a result, a vicious cycle of immune system activation and aberrant vascular inflammation is installed, ending up in target-organ damage and the further progression of EH [12] (Figure 1).
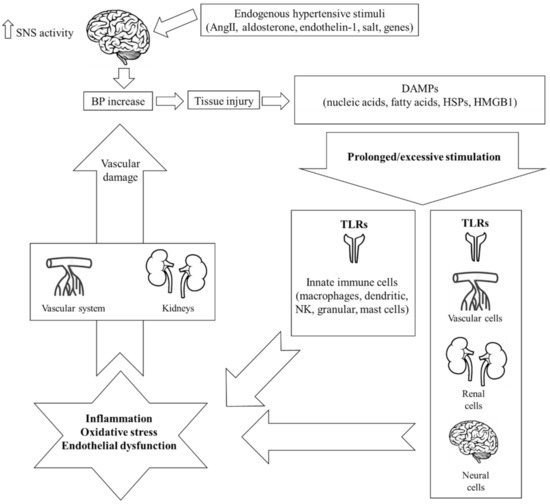
Whereas data support the participation of adaptive immunity in the pathogenesis of hypertension [16][18], the extent of the involvement of innate immunity, the precise mechanisms that stimulate it as well as its subsequent firing effects on adaptive immunity are still not well understood. To this end, the key role of TLRs in stimulating innate immunity and promoting inflammation in the hypertensive environment, merits further investigation.
4. Toll-Like Receptors
TLRs are type I transmembrane proteins belonging to the class of pattern recognition receptors (PRRs). In general, PRRs are in charge of recognizing and initiating an inflammatory reaction in response to unique, evolutionary conserved motifs termed either pathogen-associated molecular patterns (PAMPs), which are produced from viral and bacterial products, or DAMPs, which are released in the context of tissue damage, cellular stress, or cell death [10][19]. To date, 10 TLR subtypes have been identified in humans [TLR (1–10)] classified into two subfamilies according to their localization—the cell surface TLRs (1, 2, 4–6), expressed on the cell surface, and the intracellular TLRs (3, 7–10), localized in the endosomal compartment [10].
TLRs constitute the primary and most crucial step in the initiation of the inflammatory response by innate immunity. Upon activation by a specific PAMP or DAMP, TLRs fire an intracellular signal transduction cascade through two different pathways, namely (i) the myeloid differentiation primary response protein 88 (MyD88)-dependent pathway, which induces the activation of the early phase nuclear factor-κB (NF-κB), and (ii) the myD88-independent pathway (Toll/interleukin-1 receptor domain-containing adaptor protein inducing interferon-b [TRIF] dependent), which induces the activation of the late phase NF-κB. Both pathways culminate in the production and release of proinflammatory cytokines, chemokines, and several co-stimulatory factors all of which facilitate the inflammatory response [10][20][21]. In the hypertensive environment, TLRs are subject to excessive or prolonged DAMP-mediated stimulation that leads to an exaggerated innate immune inflammatory response and vascular damage.
5. The Role of Toll-Like Receptors in the Pathogenesis of Hypertension
A concise amount of data has demonstrated evidence of increased TLR activation leading to inflammation in hypertension [8][9][10][22]. Importantly, enhanced TLR expression has been documented not only on immune cells but also on several non-immune cells across the renal, vascular, and neural tissues, representing the three most vital target organs involved in the pathogenesis of EH (Table 1).
Table 1. Experimental populations where TLR activation has been documented and the relevant subtypes of TLRs involved in the pathogenesis of hypertension according to organ system.
Organ System | TLR Subtype | Experimental Populations | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Central Nervous System | TLR4 | TLR7 | TLR8 | TLR9 | Normotension [23] | Pre-hypertensive SHRs [24] | SHRs [ | AngII-induced hypertension [ ][28] | ||||||||||||
Vascular System | TLR2 | TLR4 | TLR9 | Normotension [29] | SHRs | AngII-induced hypertension [ 33] | L-NAME induced hypertension [ ] | |||||||||||||
Renal System | TLR4 | AngII-induced hypertension [35] | Aldosterone-induced hypertension [36] |
AngII: angiotensin II; L-NAME: NG-nitro-L-arginine methyl ester; SHRs: spontaneously hypertensive rats; TLR: toll-like receptor.
References
- Stephen S Lim; Theo Vos; Abraham D Flaxman; Goodarz Danaei; Kenji Shibuya; Heather Adair-Rohani; Mohammad A AlMazroa; Markus Amann; H Ross Anderson; Kathryn G Andrews; et al.Martin AryeeCharles AtkinsonLoraine J BacchusAdil N BahalimKalpana BalakrishnanJohn BalmesSuzanne Barker-ColloAmanda BaxterMichelle L BellJed D BloreFiona BlythCarissa BonnerGuilherme BorgesRupert BourneMichel BoussinesqMichael BrauerPeter BrooksNigel G BruceBert BrunekreefClaire Bryan-HancockChiara BucelloRachelle BuchbinderFiona BullRichard T BurnettTim E ByersBianca CalabriaJonathan CarapetisEmily CarnahanZoe ChafeFiona CharlsonHonglei ChenJian Shen ChenAndrew Tai-Ann ChengJennifer Christine ChildAaron CohenK Ellicott ColsonBenjamin C CowieSarah DarbySusan DarlingAdrian DavisLouisa DegenhardtFrank DentenerDon C Des JarlaisKaren DevriesMukesh DheraniEric DingE Ray DorseyTim DriscollKaren EdmondSuad Eltahir AliRebecca E EngellPatricia J ErwinSaman FahimiGail FalderFarshad FarzadfarAlize FerrariMariel M FinucaneSeth FlaxmanFrancis Gerry R FowkesGreg FreedmanMichael K FreemanEmmanuela GakidouSantu GhoshEdward GiovannucciGerhard GmelKathryn GrahamRebecca GraingerBridget GrantDavid GunnellHialy R GutierrezWayne HallHans W HoekAnthony HoganH Dean HosgoodDamian HoyHoward HuBryan J HubbellSally J HutchingsSydney E IbeanusiGemma L JacklynRashmi JasrasariaJost B JonasHaidong KanJohn A KanisNicholas KassebaumNorito KawakamiYoung-Ho KhangShahab KhatibzadehJon-Paul KhooCindy KokFrancine LadenRatilal LallooQing LanTim LathleanJanet L LeasherJames LeighYang LiJohn Kent LinSteven E LipshultzStephanie LondonRafael LozanoYuan LuJoelle MakReza MalekzadehLeslie MallingerWagner MarcenesLyn MarchRobin MarksRandall MartinPaul McGaleJohn McGrathSumi MehtaZiad A MemishGeorge A MensahTony R MerrimanRenata MichaCatherine MichaudVinod MishraKhayriyyah Mohd HanafiahAli A MokdadLidia MorawskaDariush MozaffarianTasha MurphyMohsen NaghaviBruce NealPaul K NelsonJoan Miquel NollaRosana NormanCasey OlivesSaad B OmerJessica OrchardRichard OsborneBart OstroAndrew PageKiran D PandeyCharles Dh ParryErin PassmoreJayadeep PatraNeil PearcePamela M PelizzariMax PetzoldMichael R PhillipsDan PopeC Arden PopeJohn PowlesMayuree RaoHomie RazaviEva A RehfuessJürgen T RehmBeate RitzFrederick P RivaraThomas RobertsCarolyn RobinsonJose A Rodriguez-PortalesIsabelle RomieuRobin RoomLisa C RosenfeldAnanya RoyLesley RushtonJoshua A SalomonUchechukwu SampsonLidia Sanchez-RieraElla SanmanAmir SapkotaSoraya SeedatPeilin ShiKevin ShieldRupak ShivakotiGitanjali M SinghDavid A SleetEmma SmithKirk R SmithNicolas Jc StapelbergKyle SteenlandHeidi StöcklLars Jacob StovnerKurt StraifLahn StraneyGeorge D ThurstonJimmy H TranRita Van DingenenAaron van DonkelaarJ Lennert VeermanLakshmi VijayakumarRobert WeintraubMyrna WeissmanRichard A WhiteHarvey WhitefordSteven T WiersmaJames D WilkinsonHywel C WilliamsWarwick WilliamsNicholas WilsonAnthony WoolfPaul YipJan M ZielinskiAlan D LopezChristopher Jl MurrayMajid Ezzati A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. The Lancet 2012, 380, 2224-2260, 10.1016/s0140-6736(12)61766-8.
- Bryan Williams; Giuseppe Mancia; Wilko Spiering; Enrico Agabiti Rosei; Michel Azizi; Michel Burnier; Denis L Clement; Antonio Coca; Giovanni De Simone; Anna Dominiczak; et al.Thomas KahanFelix MahfoudJosep RedonLuis RuilopeAlberto ZanchettiMary KerinsSverre E KjeldsenReinhold KreutzStephane LaurentGregory Y H LipRichard McManusKrzysztof NarkiewiczFrank RuschitzkaRoland E SchmiederEvgeny ShlyakhtoCostas TsioufisVictor AboyansIleana DesormaisGuy De BackerAnthony M HeagertyStefan AgewallMurielle BochudClaudio BorghiPierre BoutouyrieJana BrguljanHéctor BuenoEnrico G CaianiBo CarlbergNeil ChapmanRenata CífkováJohn G F ClelandJean-Philippe ColletIoan Mircea ComanPeter W De LeeuwVictoria DelgadoPaul DendaleHans-Christoph DienerMaria DorobantuRobert FagardCsaba FarsangMarc FerriniIan M GrahamGuido GrassiHermann HallerF D Richard HobbsBojan JelakovicCatriona JenningsHugo A KatusAbraham A KroonChristophe LeclercqDragan LovicEmpar LurbeAthanasios J ManolisTheresa A McDonaghFranz MesserliMaria Lorenza MuiesanUwe NixdorffMichael Hecht OlsenGianfranco ParatiJoep PerkMassimo Francesco PiepoliJorge PoloniaPiotr PonikowskiDimitrios J RichterStefano F RimoldiMarco RoffiNaveed SattarPetar M SeferovicIain A SimpsonMiguel Sousa-UvaAlice V StantonPhilippe Van De BornePanos VardasMassimo VolpeSven WassmannStephan WindeckerJose Luis ZamoranoEmanuele BarbatoVeronica DeanDonna FitzsimonsOliver GaemperliGerhard HindricksBernard IungPeter JüniJuhani KnuutiPatrizio LancellottiAndrzej JanuszewicsSalim BenkheddaParounak ZelveianPeter SiostrzonekRuslan NajafovOlga PavlovaMichel De PauwLarisa Dizdarevic-HudicDimitar RaevNikos KarpettasAleš LinhartAmin Fouad ShakerMargus ViigimaaKaj MetsärinneMarija VavlukisJean-Michel HalimiZurab PagavaHeribert SchunkertCostas ThomopoulosDénes PállKarl AndersenMichael ShechterGiuseppe MercuroGani BajraktariTatiana RomanovaKārlis TrušinskisGeorges A SaadeGintare SakalyteStéphanie NoppeDaniela Cassar DeMarcoAlexandru CarausJanneke WittekoekTonje Amb AksnesPiotr JankowskiDragos VinereanuElena I BaranovaMarina FoscoliAna Djordjevic DikicSlavomira FilipovaZlatko FrasVicente Bertomeu-MartínezThilo BurkardWissem SdiriSinan AydogduYuriy SirenkoAdrian BradyThomas WeberIrina LazarevaTine De BackerSekib SokolovicJiri WidimskyIlkka PörstiThierry DenolleBernhard K KrämerGeorge S StergiouMarius MiglinasEva GerdtsAndrzej TykarskiManuel De Carvalho RodriguesIrina ChazovaJulian SeguraAnders GottsäterAntoinette Pechère-BertschiSerap ErdineESC Scientific Document Group 2018 ESC/ESH Guidelines for the management of arterial hypertension. European Heart Journal 2018, 39, 3021-3104, 10.1093/eurheartj/ehy339.
- Katherine T. Mills; Joshua D. Bundy; Tanika N. Kelly; Jennifer E. Reed; Patricia M. Kearney; Kristi Reynolds; Jing Chen; Jiang He; Global Disparities of Hypertension Prevalence and Control. Circulation 2016, 134, 441-450, 10.1161/circulationaha.115.018912.
- Mohammad H. Forouzanfar; Patrick Liu; Gregory A. Roth; Marie Ng; Stan Biryukov; Laurie Marczak; Lily Alexander; Kara Estep; Kalkidan Hassen Abate; Tomi F. Akinyemiju; et al.Raghib AliNelson Alvis-GuzmanPeter AzzopardiAmitava BanerjeeTill BärnighausenArindam BasuTolesa BekeleDerrick A. BennettSibhatu BiadgilignFerrán Catalá-LópezValery L. FeiginJoao C. FernandesFlorian FischerAlemseged Aregay GebruPhilimon GonaRajeev GuptaGraeme J. HankeyJost B. JonasSuzanne E. JuddYoung-Ho KhangArdeshir KhosraviYun Jin KimRuth W. KimokotiYoshihiro KokuboDhaval KolteAlan LopezPaulo A. LotufoReza MalekzadehYohannes Adama MelakuGeorge A. MensahAwoke MisganawAli H. MokdadAndrew E. MoranHaseeb NawazBruce NealFrida Namnyak NgalesoniTakayoshi OhkuboFarshad PourmalekAnwar RafayRajesh Kumar RaiDavid Rojas-RuedaUchechukwu K. SampsonItamar S. SantosMonika SawhneyAletta E. SchutteSadaf SepanlouGirma Temam ShifaIvy ShiueBemnet Amare TedlaAmanda G. ThriftMarcello TonelliThomas TruelsenNikolaos TsilimparisKingsley Nnanna UkwajaOlalekan UthmanTommi VasankariNarayanaswamy VenketasubramanianVasiliy Victorovich VlassovTheo VosRonny WestermanLijing L. YanYuichiro YanoNaohiro YonemotoMaysaa El Sayed ZakiChristopher J. L. Murray Global Burden of Hypertension and Systolic Blood Pressure of at Least 110 to 115 mm Hg, 1990-2015. JAMA 2017, 317, 165-182, 10.1001/jama.2016.19043.
- Suzanne Oparil; Maria Czarina Acelajado; George L. Bakris; Dan R. Berlowitz; Renata Cífková; Anna F. Dominiczak; Guido Grassi; Jens Jordan; Neil R. Poulter; Anthony Rodgers; et al.Paul K. Whelton Hypertension. Nature Reviews Disease Primers 2018, 4, 1-21, 10.1038/nrdp.2018.14.
- Allison E. Norlander; Meena S. Madhur; David G. Harrison; The immunology of hypertension. Journal of Experimental Medicine 2017, 215, 21-33, 10.1084/jem.20171773.
- Yu Wang; Erfei Song; Bo Bai; Paul M. Vanhoutte; Toll-like receptors mediating vascular malfunction: Lessons from receptor subtypes. Pharmacology & Therapeutics 2016, 158, 91-100, 10.1016/j.pharmthera.2015.12.005.
- Cameron G. McCarthy; Styliani Goulopoulou; Camilla F. Wenceslau; Kathryn Spitler; Takayuki Matsumoto; R. Clinton Webb; Toll-like receptors and damage-associated molecular patterns: novel links between inflammation and hypertension. American Journal of Physiology-Heart and Circulatory Physiology 2014, 306, H184-H196, 10.1152/ajpheart.00328.2013.
- Gisele F. Bomfim; Fernanda Luciano Rodrigues; Fernando S. Carneiro; Are the innate and adaptive immune systems setting hypertension on fire?. Pharmacological Research 2017, 117, 377-393, 10.1016/j.phrs.2017.01.010.
- Styliani Goulopoulou; Cameron G. McCarthy; R. Clinton Webb; Toll-like Receptors in the Vascular System: Sensing the Dangers Within. Pharmacological Reviews 2015, 68, 142-167, 10.1124/pr.114.010090.
- Antoine Caillon; Ernesto L. Schiffrin; Role of Inflammation and Immunity in Hypertension: Recent Epidemiological, Laboratory, and Clinical Evidence. Current Hypertension Reports 2016, 18, 1-9, 10.1007/s11906-016-0628-7.
- William G. McMaster; Annet Kirabo; Meena S. Madhur; David G. Harrison; Inflammation, Immunity, and Hypertensive End-Organ Damage. Circulation Research 2015, 116, 1022-1033, 10.1161/circresaha.116.303697.
- Jean S. Marshall; Richard Warrington; Wade Watson; Harold L. Kim; An introduction to immunology and immunopathology. Allergy, Asthma & Clinical Immunology 2018, 14, 1-10, 10.1186/s13223-018-0278-1.
- Irene Gavras; Athanasios J. Manolis; Haralambos Gavras; The α2-adrenergic receptors in hypertension and heart failure: experimental and clinical studies. Journal of Hypertension 2001, 19, 2115-2124, 10.1097/00004872-200112000-00001.
- Haidong Zhu; Joseph Poole; Yanhui Lu; Gregory A Harshfield; Frank A Treiber; Harold Snieder; Yanbin Dong; Sympathetic Nervous System, Genes and Human Essential Hypertension. Current Neurovascular Research 2005, 2, 303-317, 10.2174/156720205774322575.
- Noureddine Idris-Khodja; Muhammad Oneeb Rehman Mian; Pierre Paradis; Ernesto L. Schiffrin; Dual opposing roles of adaptive immunity in hypertension. European Heart Journal 2014, 35, 1238-1244, 10.1093/eurheartj/ehu119.
- Grant R. Drummond; Antony Vinh; Tomasz J. Guzik; Christopher G. Sobey; Immune mechanisms of hypertension. Nature Reviews Immunology 2019, 19, 517-532, 10.1038/s41577-019-0160-5.
- Tomasz P. Mikolajczyk; Tomasz J. Guzik; Adaptive Immunity in Hypertension. Current Hypertension Reports 2019, 21, 1-12, 10.1007/s11906-019-0971-6.
- Osamu Takeuchi; Shizuo Akira; Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805-820, 10.1016/j.cell.2010.01.022.
- Takumi Kawasaki; Taro Kawai; Toll-Like Receptor Signaling Pathways. Frontiers in Immunology 2014, 5, 461-461, 10.3389/fimmu.2014.00461.
- Stavroula Ntoufa; Maria Giovanna Vilia; Kostas Stamatopoulos; Paolo Ghia; Marta Muzio; Toll-like receptors signaling: A complex network for NF-κB activation in B-cell lymphoid malignancies. Seminars in Cancer Biology 2016, 39, 15-25, 10.1016/j.semcancer.2016.07.001.
- Cameron G. McCarthy; Styliani Goulopoulou; R. Clinton Webb; Paying the Toll for Inflammation. Hypertension 2019, 73, 514-521, 10.1161/hypertensionaha.118.11782.
- Gustavo S. Masson; Anand R. Nair; Rahul B. Dange; Pedro Paulo Silva-Soares; Lisete C. Michelini; Joseph Francis; Toll-Like Receptor 4 Promotes Autonomic Dysfunction, Inflammation and Microglia Activation in the Hypothalamic Paraventricular Nucleus: Role of Endoplasmic Reticulum Stress. PLOS ONE 2015, 10, e0122850, 10.1371/journal.pone.0122850.
- Sailesh C. Harwani; Mark W. Chapleau; Kevin L. Legge; Zuhair K. Ballas; François M. Abboud; Neurohormonal Modulation of the Innate Immune System Is Proinflammatory in the Prehypertensive Spontaneously Hypertensive Rat, a Genetic Model of Essential Hypertension. Circulation Research 2012, 111, 1190-1197, 10.1161/circresaha.112.277475.
- Rahul B Dange; Deepmala Agarwal; Ryoichi Teruyama; Joseph Francis; Toll-like receptor 4 inhibition within the paraventricular nucleus attenuates blood pressure and inflammatory response in a genetic model of hypertension. Journal of Neuroinflammation 2015, 12, 31-15, 10.1186/s12974-015-0242-7.
- Hong-Bao Li; Xiang Li; Chan-Juan Huo; Qing Su; Jing Guo; Zu-Yi Yuan; Guo-Qing Zhu; Xiao-Lian Shi; Jin-Jun Liu; Yu-Ming Kang; et al. TLR4/MyD88/NF-κB signaling and PPAR-γ within the paraventricular nucleus are involved in the effects of telmisartan in hypertension. Toxicology and Applied Pharmacology 2016, 305, 93-102, 10.1016/j.taap.2016.06.014.
- Zufeng Ding; Shijie Liu; Xianwei Wang; Magomed Khaidakov; Yubo Fan; Xiaoyan Deng; David Xiang; Jawahar L. Mehta; Lectin-like oxidized low-density lipoprotein receptor-1 regulates autophagy and Toll-like receptor 4 in the brain of hypertensive mice. Journal of Hypertension 2015, 33, 525-533, 10.1097/hjh.0000000000000411.
- Rahul B. Dange; Deepmala Agarwal; Gustavo S. Masson; Jorge Vila; Brad Wilson; Anand Nair; Joseph Francis; Central blockade of TLR4 improves cardiac function and attenuates myocardial inflammation in angiotensin II-induced hypertension. Cardiovascular Research 2014, 103, 17-27, 10.1093/cvr/cvu067.
- Cameron G. McCarthy; Camilla F. Wenceslau; Styliani Goulopoulou; Safia Ogbi; Babak Baban; Jennifer C. Sullivan; Takayuki Matsumoto; R. Clinton Webb; Circulating mitochondrial DNA and Toll-like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovascular Research 2015, 107, 119-130, 10.1093/cvr/cvv137.
- Priscila R. De Batista; Roberto Palacios; Angela Martín; Raquel Hernanz; Cindy T. Médici; Marito A. S. C. Silva; Emilly M. Rossi; Andrea Aguado; Dalton V. Vassallo; Mercedes Salaices; et al.María J. Alonso Toll-Like Receptor 4 Upregulation by Angiotensin II Contributes to Hypertension and Vascular Dysfunction through Reactive Oxygen Species Production. PLOS ONE 2014, 9, e104020, 10.1371/journal.pone.0104020.
- Gisele F. Bomfim; Rosangela A. Dos Santos; Maria Aparecida Oliveira; Fernanda R. Giachini; Eliana H. Akamine; Rita C. Tostes; Zuleica B. Fortes; R. Clinton Webb; Maria Helena C. Carvalho; Toll-like receptor 4 contributes to blood pressure regulation and vascular contraction in spontaneously hypertensive rats. Clinical Science 2012, 122, 535-543, 10.1042/cs20110523.
- G.F. Bomfim; C. Echem; C.B. Martins; T.J. Costa; S.M. Sartoretto; R.A. Dos Santos; M.A. Oliveira; E.H. Akamine; Z.B. Fortes; R.C. Tostes; et al.R.C. WebbM.H.C. Carvalho Toll-like receptor 4 inhibition reduces vascular inflammation in spontaneously hypertensive rats. Life Sciences 2015, 122, 1-7, 10.1016/j.lfs.2014.12.001.
- R. Hernanz; S. Martínez-Revelles; R. Palacios; A. Martín; V. Cachofeiro; A. Aguado; L. García-Redondo; M. T. Barrús; P. R. De Batista; A. M. Briones; et al.M. SalaicesM. J. Alonso Toll-like receptor 4 contributes to vascular remodelling and endothelial dysfunction in angiotensin II-induced hypertension. British Journal of Pharmacology 2015, 172, 3159-3176, 10.1111/bph.13117.
- Daniel Sollinger; Ruth Eißler; Steffen Lorenz; Susanne Strand; Stefan Chmielewski; Cristiane Aoqui; Christoph Schmaderer; Hans Bluyssen; Josef Zicha; Oliver Witzke; et al.Elias SchererJens LutzUwe HeemannMarcus Baumann Damage-associated molecular pattern activated Toll-like receptor 4 signalling modulates blood pressure in l-NAME-induced hypertension. Cardiovascular Research 2013, 101, 464-472, 10.1093/cvr/cvt265.
- Sathnur Pushpakumar; Lu Ren; Sourav Kundu; Alejandra Gamon; Suresh C. Tyagi; Utpal Sen; Toll-like Receptor 4 Deficiency Reduces Oxidative Stress and Macrophage Mediated Inflammation in Hypertensive Kidney. Scientific Reports 2017, 7, 1-15, 10.1038/s41598-017-06484-6.
- Yide Zhang; Weisheng Peng; Xiang Ao; Houyong Dai; Li Yuan; Xinzhong Huang; Qiaoling Zhou; TAK-242, a Toll-Like Receptor 4 Antagonist, Protects against Aldosterone-Induced Cardiac and Renal Injury. PLOS ONE 2015, 10, e0142456, 10.1371/journal.pone.0142456.
- Sathnur Pushpakumar; Lu Ren; Sourav Kundu; Alejandra Gamon; Suresh C. Tyagi; Utpal Sen; Toll-like Receptor 4 Deficiency Reduces Oxidative Stress and Macrophage Mediated Inflammation in Hypertensive Kidney. Scientific Reports 2017, 7, 1-15, 10.1038/s41598-017-06484-6.
- Yide Zhang; Weisheng Peng; Xiang Ao; Houyong Dai; Li Yuan; Xinzhong Huang; Qiaoling Zhou; TAK-242, a Toll-Like Receptor 4 Antagonist, Protects against Aldosterone-Induced Cardiac and Renal Injury. PLOS ONE 2015, 10, e0142456, 10.1371/journal.pone.0142456.