The chemistry of selenium (Se) compounds is not a young field, since its beginning was in the first half of the 19th century. The first steps in this scope were initiated in 1836 by the then toxicologists, who discovered a Se metabolite, diethyl selenide, excreted by inhalation during research using inorganic selenium compounds on animals. Meanwhile, the first synthesis of selenium-containing (Se-containing) compounds took place in 1847, when ethylselenol was obtained. Since then, selenium chemistry has developed greatly and investigators have started to look at whether it can help with society’s health problems, including cancer. The high number of new cancer incidences and the associated mortality continue to be alarming, leading to the search for new therapies that would be more effective and less burdensome for patients. As there is evidence that Se compounds can have chemopreventive activity, studies have begun to establish whether these compounds can also affect already existing cancers. This review aims to discuss the different classes of Se-containing compounds, both organic and inorganic, natural and synthetic, and the mechanisms and molecular targets of their anticancer activity.
- selenium compounds
- cancer
- anticancer agents
1. Inorganic Selenium Compounds
Both selenite (SeO32−, SeL, Figure 1, structure 1) and its sodium salt, sodium selenite (disodium selenite, Na2SeO3, SS, Figure 1, structure 2), belong to a group of inorganic compounds, which were tested as the first Se-containing compounds, and the scope of research on their anticancer properties was very extensive [1][2][3].
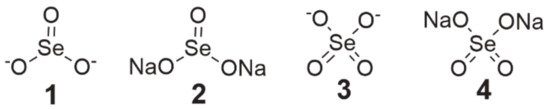
Figure 1. Chemical structure of inorganic selenium compounds (1—selenite, 2—sodium selenite, 3—selenate, and 4—sodium selenate).
During the metabolism of SS in vivo, the formation of hydrogen selenide (H2Se) occurs [4][5], which is then methylated forming methylselenol [6][7]. The in vivo study concluded that SeL (3 mg/kg body weight (b.w.)) was well tolerated but was genotoxic [8], while at a concentration of about 127 µM Se, it was toxic and genotoxic to primary human keratinocytes (NHK) [9]. This is due to the fact that medium (3–5 ppm; 0.3–0.5 mg Se/kg b.w.) or dietary (0.1 ppm; 0.01 mg Se/kg b.w.) doses of SeL are metabolized to selenide (Se2−), which is then incorporated into selenoproteins [1][10][11]. In turn, higher concentrations (>5 ppm; >0.5 mg Se/kg b.w.) of this compound lead to its reaction with oxygen and the formation of ROS, which causes oxidative stress, and this ultimately results in the cytotoxic and genotoxic effects [1][9][11][12]. Selenate (Figure 1, structure 3) also exhibits genotoxic properties [8]. For this reason, chronic high doses of SeL should not be ingested, as its genotoxic properties in normal cells may lead to their mutagenesis and paradoxical cancer development [8]. It is worth quoting here the results of a study in which a combination of SeL (126.6 µM Se) and Trolox (80 µM; water-soluble Vit. E) increased the prooxidative and genotoxic effects of SeL on NHK cells, which could have resulted from the formation of vitamin E radicals by SeL. No such results were observed when using selenomethionine (SeMet) + Trolox, which proves that SeL is a prooxidant [9]. The anticancer effects of SeL and SS were observed on many cancer cell lines, including prostate [5][6][7], breast [13], lung [14], hepatoma [15], bladder [16], and glioblastoma [17] or osteosarcoma [18].
Furthermore, cells resistant to the available cytostatic agents in comparison with cells sensitive to them are more susceptible to SeL [2]. It was reported that SeL affects and disrupts intracellular GSH levels. This is because SeL is reduced to selenide (Se2−) by GSH, resulting in the rapid depletion of GSH in the cell [15]. The selenide produced as a result of this reaction reacts with oxygen, leading to ROS formation [11]. Since GSH is too low in the cell, there is a redox imbalance and excessive amounts of ROS form, which in turn induces oxidative stress [11][14][15][16] and other signal pathways, and leads to apoptosis [3]. In a study by Shigemi et al. [19], excessive amounts of ROS caused ER stress and subsequent apoptosis via activating the unfolded protein response (UPR) pathway as a result of SS activity.
However, in the apoptosis induced by SeL, mitochondria are the main contributor [3][15]. SS easily diffuses into the mitochondria [11], reacting with the mitochondrial GSH and producing superoxide, which reacts with the components of this cellular organelle [20]. Then, the oxidation of protein thiol groups occurs. Both the generated superoxides and the oxidized thiols open the pores of the mitochondrial permeability transition (MPT) [15][20]. At this point, the potential of the mitochondrial membrane (ΔΨm) decreases [11][15][16][20] and cytochrome c is released into the cytoplasm [1][20]. In the case of selenate, this apoptotic pathway does not occur [11][20].
In addition, the p53 protein is involved in the apoptotic pathway. As SeL leads to DNA fragmentation (DNA fragments 50–300 bp) and DNA SSBs [1], a signal of DNA damage appears, which leads to p53 activation [5][12]. The activation of p53 increases the levels of the proapoptotic protein, Bax [5][18], which is then translocated into the mitochondria, oligomerizes, and finally destabilizes the mitochondrial membrane [5]. Furthermore, the introduction of cells into the apoptotic pathway may be caspase-mediated [6][7][11], but it may also be a caspase-independent process [3][5].
To sum up the above-described mechanisms, as a result of SeL metabolism, excessive generation of ROS is observed, which results in DNA SSBs and activation (phosphorylation) of p53, which in turn induces increased transcription of Bax and then its (Bax) translocation to the mitochondria. The membrane is destabilized (opening of MPT pores) and cytochrome c is released to cytosol, which activates caspases and finally leads to cancer cell apoptosis. The caspase-independent apoptosis occurs in cells without a functional p53 protein (e.g., prostate cancer PC-3 or DU145 cells), but is not as efficient as caspase-dependent apoptosis (no p53-mitochondria feedback loop, which sustains ROS production) [5]. As can be seen, apoptosis occurs through the internal (mitochondrial) pathway, but there are reports of a component of the external pathway through the DR5 death receptor. This was observed in DU145 prostate cancer cells, where SeL induced the upregulation of the DR5 receptor and simultaneous activation of the internal pathway, which may lead to the assumption that the internal and external pathways may be related [1].
Moreover, the inhibition of cancer cell proliferation by SeL may occur by suppressing NF-κB DNA binding [6], inhibiting protein kinase C (PKC) [3][10] or histone deacetylases (HDAC) [2][13] activity, as well as poisoning topoisomerase II (by reacting with thiols incorporated into this enzyme) [12] or by affecting other molecular targets, i.e., p38 mitogen-activated protein kinase (p38MAPK), c-Jun NH2-terminal kinase (JNK), AP-1 [1][3][7], ATM [12], AKT [3][7]. As far as the cell cycle is concerned, SeL treatment led to cell cycle arrest in phase S and irreversible inhibition of cell proliferation, while selenate arrested the cycle in phase G2, and growth inhibition was reversible [7][11]. So far, the impact of SeL on apoptotic events in cancer cells has been described. Apart from the apoptotic phenotype, there are several other non-apoptotic types of cell death, i.e., necrotic or autophagic phenotype, which had also been observed during SeL treatment [3]. In the study carried out by de Miranda et al. [13], the authors observed that SeL induced necrosis in breast cancer MCF-7 cells, while Soukupová et al. [16] found autophagy and necrosis in chemoresistant human bladder cancer RT-112/D21 cells. In addition, Subburayan et al. [21] reported in their paper that SS induced ferroptosis in human cancer cells such as breast (MCF-7), prostate (PC-3), and glioma (U-87MG).
In addition to the research on the effect of SeL on cancer cells, there were also studies that used combination therapy of SeL with standard chemotherapy. Thus, it was proved that SeL synergistically enhanced the effectiveness of imatinib against colorectal cancer HTC116, cisplatin against MCF-7 cells [22], or TrxR (thioredoxin reductase) inhibitors (auranofine, ethaselen) against human lung and ovarian cancer cells [2]. Furthermore, it was reported that apart from synergistic enhancement, SeL also reduced the side effects (e.g., toxicity) of commonly used cytostatic drugs, among other (a.o.) 5-fluorouracil (5-FU) or docetaxel [2]. There are also human studies, in which the combination of SS with commonly used cancer therapies was applied. Table 1 shows clinical trials of such combined therapies and the study using SS alone.
Table 1. Sodium selenite under clinical investigation. This table is compiled from the information available at https://www.clinicaltrials.gov/ on 7 January 2021.
| NCT Number | Study Title | Clinical Trial Status | Study Design | References |
|---|---|---|---|---|
| NCT04296578 | Study evaluating sodium selenite in combination with abiraterone in castration-resistant prostate cancer progressing on abiraterone | Phase 1 not yet recruiting |
Non-randomized, open-label, single-group assignment | [23] |
| NCT01155791 | Phase I sodium selenite in combination with docetaxel in castration-resistant prostate cancer | Phase 1 terminated |
Open-label, single-group assignment | [24] |
| NCT02184533 | Sodium selenite and radiation therapy in treating patients with metastatic cancer | Phase 1 completed |
Open-label, single-group assignment | [25] |
| NCT00188604 | The use of selenium to treat secondary lymphedema breast cancer | Phase 2 completed |
Randomized, double-blind, placebo-controlled, crossover assignment | [26] |
| NCT01959438 | Sodium selenite as a cytotoxic agent in advanced carcinoma (SECAR) | Unknown | Open-label, single-group assignment | [27] |
| NCT04201561 | High dose inorganic selenium for preventing chemotherapy induced peripheral neuropathy (SELENIUM) | Phase 3 recruiting |
Randomized, triple-blind (participant, care provider, investigator), placebo-controlled, parallel assignment | [28] |
In conclusion, during the use of high doses of SeL, the selenide (Se2−) formed during metabolism is responsible for the genotoxic effects resulting from the formation of large amounts of ROS, which interact with DNA. On the other hand, methylation of Se2− leads to obtaining a compound (methylselenol) that does not show this property. This has led to the design of stable compounds, which are precursors (“prodrugs”), releasing anticancer methylselenol during their metabolism [6]. These methylselenol precursors will be discussed in the following section of this review.
2. Organic Selenium Compounds
2.1. Diselenides
Diselenides are a class of compounds that contain the Se-Se bond in their structure and have the general chemical formula R2Se2. Dimethyl diselenide (Figure 2, structure 5) is a compound found in nature that exhibits antioxidant properties and strongly induces NADPH quinone oxidoreductase, whereas dipropyl diselenide (Figure 2, structure 6) and dibutyl diselenide (Figure 2, structure 7) are known for their prooxidant effects, even at low concentrations [29][30].
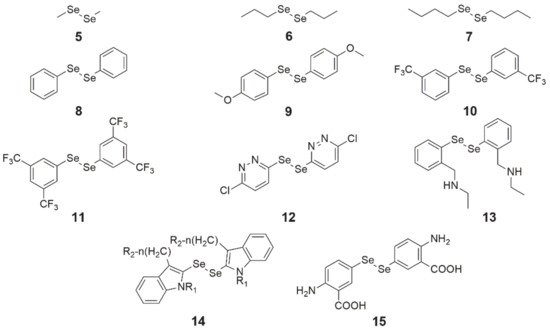
Figure 2. Chemical structures of diselenide derivatives (5—dimethyl diselenide, 6—dipropyl diselenide, 7—dibutyl diselenide, 8—diphenyl diselenide, 9—4,4′-dimethoxy-diphenyl diselenide, 10—3,3′-ditrifluoromethyl-diphenyl diselenide, 11—3′,5′,3,5-tetratrifluoromethyl-diphenyl diselenide, 12—1,2-bis(chloropyridazinyl) diselenide, 13—N,N’-((diselanediylbis(2,1-phenylene))bis(methylene))di(ethaneamine), 14—indol-containing diselenide, 15—bis(4-amino-3-carboxyphenyl) diselenide).
In the case of diphenyl diselenide (Figure 2, structure 8), it has been characterized as an antioxidant and inhibitor of nociception, which also protects neurons from damage and has antidepressant properties [31]. Its protective activity on cells was tested on murine J744 macrophage-like cells and during tamoxifen (TAM) therapy on mammary gland cancer MCF-7 cells. In the first case, after the generation of ROS and subsequent treatment with compound 8, there was a decrease in NF-κB expression in the cells, while in the second study, it counteracted oxidative damage in cells caused by TAM without affecting its activity. At low doses, it had protective properties against genotoxic substances.
Meanwhile, in vivo studies revealed that diphenyl diselenide can be toxic, depending on its route of administration and dosage [29]. Mainly, this compound was studied for its antioxidant activity, while there is less research on its cytotoxic effects on cancer cells. The inhibitory activity of compound 8 on neuroblastoma (SH-SY5Y) cell growth seems to be mediated by the extracellular signal-regulated kinase (ERK 1/2) pathway, which was presented in a paper by Posser et al. [32]. Additionally, in the study by Nedel et al. [31], compound 8 at a concentration of 80 µM exhibited cytotoxicity and apoptosis induction in HT-29 cells, which is consistent with the findings of Posser et al. [32] that diphenyl diselenide leads to cell death via the apoptosis pathway.
In turn, 4,4′-dimethoxy- and 3,3′-ditrifluoromethyl-diphenyl diselenide (Figure 2, structures 9 and 10) were more cytotoxic than 8 to HT-29 cells and induced apoptosis at 20 µM, out of which 9 caused a higher percentage of this cellular event. It was found that compounds 9 and 10 induced apoptosis via the caspase-dependent (↑ AIF, ↑ caspase 9 and 8) and caspase-independent (↑ Bax, ↓ survivin) pathways, and arrested the cell cycle in the G2/M phase via genes p53, p21 (↑ expression), and MYC (↓ expression) [31].
Studies on the cytotoxicity of 3′,5′,3,5-tetratrifluoromethyl-diphenyl diselenide (Figure 2, structure 11) were carried out on several cancer cell lines, i.e., HL-60 (human leukemia, IC50 = 8 µM), PC-3 (prostate cancer, IC50 = 13 µM), MCF-7 (breast cancer, IC50 = 18 µM), MIA-PA-Ca-2 (pancreatic cancer, IC50 = 25 µM) and HCT-116 (colorectal cancer, IC50 = 27 µM). Compound 11 induced apoptosis via the internal pathway (mitochondrial) and arrested cells in phase S of the cell cycle. A simulation of molecular docking of diselenide 11 demonstrated that this compound selectively binds to the minor groove of DNA via hydrogen bonds in which fluorine atoms are involved. Additionally, there are hydrophobic interactions between the benzene rings and DNA.
1,2-bis(chloropyridazinyl) diselenide (Figure 2, structure 12) had the highest growth inhibition of MCF-7 cancer cells among all 17 compounds synthesized and studied by Kim et al. [33]. Proliferation inhibition by this compound was dose-dependent and its IC50 amounted to 10.34 µM.
N,N’-((diselanediylbis(2,1-phenylene))bis(methylene))di(ethaneamine) (Figure 2, structure 13), which was synthesized by Krasowska et al. [34], exhibited the inhibition of cell proliferation in micromolar concentrations in three cell lines of cervical (HeLa, IC50 = 20 µM) cancer, breast (MCF-7, IC50 = 30 µM) cancer, and leukemia (K652, IC50 = 15 µM). It was found that 13 is an inhibitor of glutathione-S-transferase (GST)—this enzyme is responsible for increased drug removal in cancer cells. Moreover, the use of this compound with cisplatin enhanced its cytotoxic activity, possibly resulting from GST inhibition. The authors also conducted a molecular docking of 13 in a crystal structure of GST in their study. It turned out that it can be located in the H-site of the active center of the enzyme and be stabilized by many interactions. It was also found that the key interactions for the inhibition of GST activity by compound 13 are π–π stacking interactions with Y108 and the diselenide bridge docked near Y7 (a phenol group).
The indol-containing diselenide (Figure 2, structure 14) and bis(4-amino-3-carboxyphenyl) diselenide (Figure 2, structure 15) affect the kinases. The activity of cyclin kinase (CDKs) is slightly suppressed by 15, while 14 inhibits the tyrosine kinase in cells [29].
In vitro studies of diselenides suggest that they may be promising candidates for anticancer agents, but their application, including, among others, that of diphenyl diselenide, may be limited due to their toxicity in vivo. The toxic effects of these compounds depend on the dosage, vehicle, and route of administration, as well as the age or species of the tested animal [35]. It was observed that diphenyl diselenide was less toxic in rats than in mice—the LD50 of this compound was 374.4 mg/kg in rats and 65.52 mg/kg in mice after intraperitoneal administration [35][36]. Acute subcutaneous (<156 mg/kg) and oral exposure to diphenyl diselenide and its chronic administration (<31.2 mg/kg, subcutaneously) did not cause toxic effects in mice. In contrast, repeated administration at >31.2 mg/kg subcutaneously and acute intraperitoneal exposure to this compound had toxic effects in these rodents [35][36][37]. Oral administration of 30 mg/kg (supranutritional dose of diphenyl diselenide) for 8 months was non-toxic in rabbits, whereas acute intraperitoneal administration of 1.56 and 15.6 mg/kg was hepatotoxic to them, while a dose of 156 mg/kg was lethal to 85% of the population [35]. The main mechanism responsible for the toxic properties of diselenides is their prooxidant activity, which involves interaction with functionally relevant -SH (thiol) groups of proteins, leading to their oxidation and loss/decrease in their function [36][38][39]. Depletion of intracellular GSH is one of these effects [36][38]. Furthermore, by the same mechanism, these compounds can inhibit the enzyme δ-aminolevulinic acid dehydratase (δ-ALA-D) in the blood, kidney, liver, and brain (highly lipophilic molecules that can cross the blood–brain barrier (BBB)) [35][36][38][39]. Inhibition of this enzyme in the blood results in the accumulation of aminolevulinic acid (ALA), characterized by some prooxidant properties [36]. In turn, the crossing of the BBB by diselenides results in their neurotoxic effects [35][39]. This is manifested by the induction of convulsions in mice after the administration of diphenyl diselenide. It is noteworthy that this effect was observed only in intraperitoneal administration, indicating that this toxicity depends on the route of administration. This may be due to the accumulation of the intermediate metabolite of diphenyl diselenide in the brain, although it seems more possible that inhibition of δ-ALA-D leads to accumulation of ALA, which has a proconvulsant effect. Interestingly, the neurotoxicity of diphenyl diselenide is also determined by the age of the tested animal. Oral administration of this compound induced seizures in rat pups, but not in adults. In addition, glutamatergic system impairment was found [35][36][39][40]. In the case of 4,4′-dimethoxy- and 3,3′-ditrifluoromethyl-diphenyl diselenide, reduction/abolition of the occurrence of convulsions in mice was observed [35][40]. The acute oral toxicity of 3,3′-ditrifluoromethyl-diphenyl diselenide was slightly higher (LD50 > 278 mg/kg) and 4,4′-dimethoxy-diphenyl diselenide was slightly lower (LD50 > 372 mg/kg) than that of diphenyl diselenide (LD50 > 312 mg/kg). This means that the introduction of substituents into the aromatic ring of diphenyl diselenide had no significant effect on reducing its toxicity. Nevertheless, 4,4′-dimethoxy-diphenyl diselenide did not inhibit δ-ALA-D and did not cause death in experimental animals—the compound with the -CF3 group exhibited both of these properties [40]. However, another member of the diselenide class, 2,2′-dithienyl diselenide, at a single oral dose of 100 mg/kg exhibited systemic toxicity in rats and caused death. In this study, inhibition of δ-ALA-D, an increase in aspartate (AST) and alanine aminotransferase (ALT) activities, and a decrease in urea levels were observed, with no effect on creatinine levels, showing that the compound had a hepatotoxic effect but no effect on renal function [39]. A very important finding is that in studies using diselenides-loaded nanocapsules, no toxic effects on mice were shown, which gives hope for a reduction in their harmful properties and possible application in human trials in the future [38][41]. Summarizing the toxicological data of diselenides in in vivo studies, more information from further animal studies on them is needed to unambiguously determine the exact mechanism of their harmful effects on the body, as well as methods to prevent them. Although the synthesis of diphenyl diselenide derivatives proved to be not entirely successful (their toxicity was comparable to that of diphenyl diselenide), the reduction in toxic effects may be probably achieved by the formulation of the administered compound.
3.2. Selenides
Another class of selenoorganic compounds is selenides (also called selenoethers), with the general formula R-Se-R. Among them, there are derivatives with chemopreventive properties, involving the binding of heavy metals or Gpx-like (glutathione peroxidase-like) activity. Meanwhile, the anticancer activity of the compounds was tested on various cancer cell lines, including colon, uterine, lung, liver, and breast cancer. Zidovudine derivatives (active at medium micromolar concentrations) induced apoptosis through the internal (mitochondrial) pathway, while phenylindolyl ketone derivative was active at nanomolar concentrations, causing the arrest of the cell cycle in the G2/M phase, which led the cells to the apoptosis pathway as a result of a decrease in mitochondrial membrane potential (ΔΨm) and additionally inhibited tubulin polymerization [29].
Another selenide compound, which is also an inhibitor of tubulin polymerization, is a combretastatin 4-A analog (Figure 3, structure 16). This analog was tested on a panel of four cancer cell lines, exhibiting an inhibitory effect on cell growth in nanomolar concentrations, and its activity was stronger than combretastatin A-4 (CA-4, Figure 3). IC50 values for this compound for breast (MCF-7), kidney (768), colon (HT-29), and prostate (PC-3) cancer were 10, 680, 280, and 80 nM, respectively. In the molecular docking simulation of compound 16, it was found that it binds at the colchicine-binding site of tubulin. The selenoether angle was 101.19°, which provides deep receptor binding in tubulin (colchicine-like). Between the oxygen atom of the methoxy substituent in the para position (ring A, Figure 3) and the thiol from the side chain of the residue b-Cys 241 of the receptor site of the enzyme, the hydrogen bond forms. Another hydrogen bond binds the oxygen atom from the para-methoxy substituent of ring B (Figure 3) with the amide nitrogen of residue a-Val181 at the receptor site of the enzyme. As a result of this mechanism, tubulin is inhibited and its enzymatic activity decreases [42].
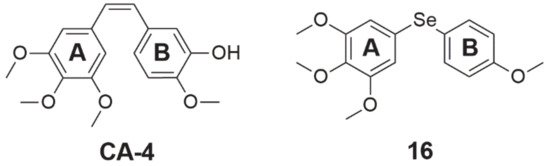
Figure 3. Chemical structures of combretastatin 4-A (CA-4) and its derivative (16).
2.3. 1,2-Benzisoselenazole-3[2H]-One Derivatives
Ebselen (2-phenyl-1,2-benzisoselenazol-3(2H)-one, 2-phenyl-1,2-benzoselenazol-3-one, also called PZ 51, DR3305, SPI-1005, Figure 4, structure 17) is a Se-containing heterocyclic compound that shows chemopreventive properties through its anti-inflammatory and antioxidant activity. This is possible due to its weak GPx-like action [1][43]. As a result of ROS scavenging, this compound prevents the development of cellular oxidative stress, protecting against the oxidation of cell components and the formation of DNA mutations [43]. Yang et al. [44] demonstrated in their study that treatment with 25 µM ebselen reduced the effect of H2O2 on growth inhibition, DNA damage, and lipid peroxidation in HepG2 cells. Among the wide range of studies on the antioxidant properties of ebselen, there are also reports of its possible inhibitory effects on proliferation. In experiments on HepG2 cells, this compound at a dose of 50–75 µM intensified the apoptosis process. This was caused by the oxidation of thiols present inside the cell, which resulted in their sharp depletion [1].
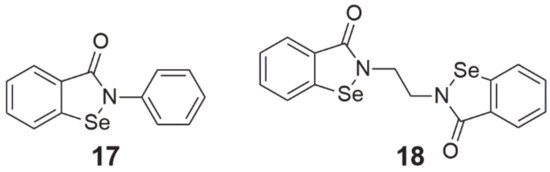
Figure 4. Chemical structures of 1,2-benzoselenazole-3[2H]-one derivatives (17—ebselen, 18—ethaselen).
In another study, ebselen inhibited the proliferation of human multiple myeloma cells, increasing their apoptosis, which was associated with high levels of ROS and their impact on mitochondria. After treating cells with 40 µM ebselen for 4 h, translocation of Bax to the mitochondria, a decrease in their membrane potential (ΔΨm), and a final release of cytochrome c into the cytoplasm were observed [43]. In pancreatic cancer xenografts in mice, ebselen at a dose of 160 and 640 µg/day (no differences in tumor size between doses) reduced tumor development by 58% [45]. In addition, other investigators showed that co-treatment of ebselen with tumor necrosis factor α (TNF-α) in glioblastoma cells resulted in increased sensitivity to TNF-α and increased apoptosis. The observed effect resulted from the activation of two different pathways, which included the deactivation of NF-κB and stimulation of death-inducing signaling complex (DISC) production, which finally led to an activation of caspase 8, triggering the executive caspase cascade and induction of apoptosis [46][47]. An in vitro study using the MCF-7 cell line showed that this compound at a concentration of 25 µg/mL combined with γ-radiation at 6 Gy resulted in the inhibition of proliferation and induction of apoptotic cell death, which resulted from the regulation of pro- and anti-inflammatory cellular response and expression of genes and proteins involved with its pathways [48].
In the case of human studies, there are two clinical trials on ebselen. Both of them are randomized, quadruple-blind (participant, care provider, investigator, outcomes assessor), placebo-controlled, and parallel assignment. The first trial (NCT number: NCT01452607) is in phase 1 and is investigating the pharmacokinetic profile of ebselen within 24 h (dose: 200 mg) and its safety (time frame: 1 month, dose range: 200–1600 mg) in healthy subjects [49]. The second clinical trial (NCT number: NCT01451853, phase 2) was designed to investigate the safety and efficacy of this compound in preventing hearing loss induced by platinum-based cytostatics (e.g., cisplatin, carboplatin). Ebselen was given orally to participants twice daily for three days of each chemotherapy cycle in three different dose schemes (low: 200 mg, middle: 400 mg, and high: 600 mg) depending on the trial arm [50].
Another compound from the 1,2-benzisoselenazole-3[2H]-one derivatives class is ethaselen (1,2-[bis(1,2-benzisoselenazolone-3(2H)-ketone)]ethane, 2-[2-(3-oxo-1,2-benzoselenazol-2-yl)ethyl]-1,2-benzoselenazol-3-one, BBSKE, Figure 4, structure 18), which seems to be a very promising molecule with potential anticancer properties [2]. This compound has been identified as a mixed-type mammalian TrxR inhibitor and its goal is the C-terminal active site of this enzyme. By binding to the redox pair Sec-Cys, it inhibits the reduction of oxidized Trx, leading to the accumulation of both ethaselen and ROS in the cell [45][51]. The suppressive effect of ethaselen on the proliferation of cancer cells has been confirmed on many cell lines in vitro and in vivo, including cancer models of the lung, tongue, stomach, liver, colon, prostate, cervix, nasopharyngeal cavity, and leukemia [3][45][51][52].
As far as the mechanism of action and the properties of this compound at the chemical level are concerned, it turns out that the place with strong electrophilicity is the bond between the selenium and nitrogen (Se-N) atoms in the benzisoselenazole ring, which affects the ability to react with -SH and -SeH groups of proteins. As a result of this reaction, diselenide (Se-Se) and selenenylsulfide (Se-S) bonds are formed between ethaselen and TrxR molecules [51]. In their work, Wang et al. [51] used the molecular docking simulation of ethaselen in the TrxR enzyme. Depending on the TrxR type, only S-Se or S-Se and Se-Se bonds could be formed. In the first stage, the nucleophilic Cys498 (Sec498 in TrxR wild type) attacks the S-N bond in the benzisoselenazole ring of ethaselen, which leads to its opening and the formation of the S-Se bond (Se-Se in the case of the wild type enzyme). In the second stage, the opening of one of the two rings increases the elasticity of the compound and causes the interaction of the S-N bond in the second ethaselene ring with Cys497 and the formation of the S-Se bond between it and the enzyme. Apart from this, the authors suggest that hydrogen bonds between His472 and Tyr116 residues and ethaselen are also involved in this mechanism, so it could affect the catalytic functions of TrxR1. As mentioned earlier, the inhibition of TrxR caused the accumulation of oxidized Trx, which resulted in a decrease in NF-κB signaling and the introduction of cells into the apoptosis pathway [52]. In addition, in Tca8113 tongue cancer cells, it was observed that as a result of TrxR inhibition, caspase 3 was activated [3].
In addition to effective ethaselen monotherapy, there is evidence that this compound increased efficacy in combination with available cytostatic drugs or radiotherapy. This compound enhanced the effectiveness of cisplatin in human lung xenografts (A549) in mice [45][52] and leukemic cells resistant to cisplatin, and acted synergistically with sunitinib against colon cancer cells [45]. In addition, it sensitized to radiotherapy non-small cell lung cancer cells (NSCLC) [46][51].
Currently, this compound is in phase 1c clinical trials (NCT number: NCT02166242) in China, where patients with non-small cell lung cancer (NSCLC) take a dose of 600 mg of ethaselen/day (safe and tolerable dose: 1200 mg/day as determined in phase 1a/b) [53].
2.4. Selenophene-Based Derivative
Among this class of compounds, great curiosity is aroused by 2,5-bis(5-hydroxymethyl-2-selenienyl)-3-hydroxymethyl-N-methylpyrrole (D-501036, Figure 5), which has a wide range of activities against some human cancer cells lines. Its effect depends on the used dose and exposure time. It was demonstrated that exposure to D-501036 caused an increase in caspase 3 and 9 activity [3]. In addition, this compound was selective in relation to cancer cells, causing apoptotic events and DNA double-strand breaks (DSBs). Its high effectiveness was observed in relation to cells exhibiting multidrug resistance (MDR) characterized by the overexpression of glycoprotein P, which gives hope for an increase in treatment efficacy in chemotherapy-resistant cancers [2].
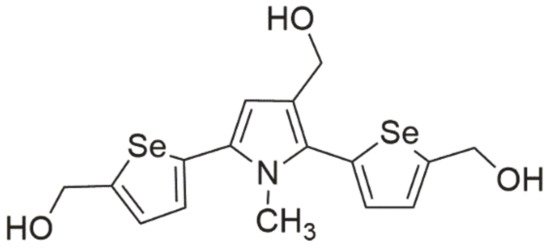
Figure 5. Selenophene-based derivative (D-501036).
2.5. Seleninic Acids
One of the most extensively investigated Se-containing compounds with anticancer properties, apart from selenite, is methylseleninic acid (methylselenic acid, methaneseleninic acid, MSA, Figure 6). It is a compound from the group of oxoacids [45], which is obtained from methylselenocysteine (MSC) during the transamination reaction or with the involvement of β-lyase, i.e., this enzyme is not necessary for MSA activity [1][2][54].
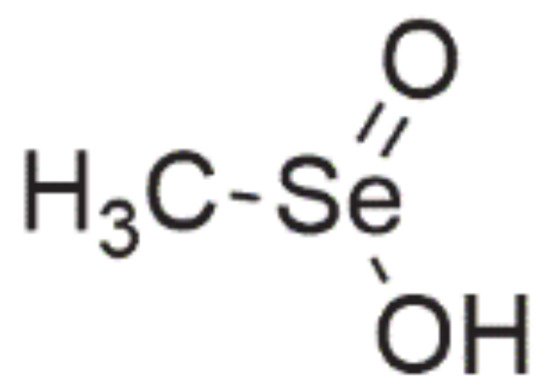
Figure 6. Chemical structure of methylseleninic acid.
In vitro, MSA has a stronger activity than MSC, but in vivo, this difference is minimized (β-lyase is present in tissues) [1]. MSA is non-genotoxic in comparison with selenite [45][8] and does not exhibit toxicity to normal body tissues [45][55][56][57]. Its impact on cancer cells was studied both in vitro and in vivo on various cell lines, i.e., prostate [5][6][7][8][54][58][59][60], colon [55], breast [13][61], liver [15], lung [14], and pancreatic [45] cancer. MSA inhibits the proliferation of cancer cells by inducing an apoptotic process via various mechanisms [45][11][15], arresting the cell cycle in phase G1 [1][7][8] and its antiangiogenic activity [55][8][11][61]. One of the possible mechanisms of MSA is the excessive generation of ROS [14].
In a study on human hepatoma HepG2 cells, it was noted that MSA (25 µM) was a strong oxidant that led to a rapid depletion of intracellular glutathione (GSH) as a result of the reaction with it and the formation of anticancer-active methylselenol [15]. It can therefore be predicted that cancer cells with increased levels of GSH in a cell will be more sensitive to the effects of MSA. As it is generally known, excessive levels of ROS in a cell cause ER stress and the induction of UPR [45]. Moreover, an important element is caspase 12 (present in ER) and its activation during ER stress, which triggers an apoptotic process in PC-3 cells [11]. The mechanism in which ER and UPR are involved was described in a study conducted by Shigemi et al. [19] using primary effusion lymphoma (PEL) cells, in which treatment with MSA increased the levels of oxidized proteins and promoted ER stress, inducing proapoptotic UPR and then leading the cell to the apoptotic pathway. In relation to the above, these results suggest that apoptosis in cancer cells may occur through a disturbed cell redox balance [11].
Meanwhile, in another study, apoptosis was ROS-independent [5]. MSA treatment of prostate cancer DU145 and PC-3 androgen-independent cells caused DNA fragmentation and caspase-dependent PARP cleavage, which indicates caspase-dependent apoptosis [4][7]. It is worth adding that apoptosis was caspase-dependent (caspase 3, 7, 8, 9), but it was not affected by p53 [45][55][5]. In a study conducted by Gasparian et al. [6], a different mechanism of anticancer action of MSA was observed. Prostate cells treated with 5 µM MSA were directed to the apoptotic pathway as a result of the inhibition of NF-κB DNA binding stimulated by TNF-α and lipopolysaccharide (LPS). This resulted from blocking the activation of the IκB kinase (IKK) by MSA and preventing the release of NF-κB. At this point, it is worth mentioning that interleukin 6 (IL-6) activates NF-κB, enabling its binding to DNA [62], because in the study carried out by Zeng et al. [55], an inhibition of proliferation in colon cancer xenografts in C57BL/6 mice as a result of treatment with MSA (3 mg/kg body weight) and a corresponding decrease in TNF-α and IL-6 levels were observed. Therefore, in this situation, it seems that NF-κB DNA binding could also be inhibited.
Summarizing these two investigations, it can be suggested that the inhibition of the inflammatory process induced by LPS, TNF-α, and IL-6 may have an impact as an anticancer and chemopreventive factor. Moreover, other MSA mechanisms of directing cancer cells to the apoptotic pathway were observed, such as the inactivation of PKC [59], inhibition of HDAC [2][13], blocking of androgen [57][60] or estrogen receptor (ER) [4] signaling, upregulation of CDK inhibitors (CDKI), which are inhibitors of kinases CDK2, -6 and -4 [3], and other molecular targets, such as the proteins regulated in development and DNA damage response 1 (REDD1), ERK 1/2, p38MAPK, JNK 1/2 or protein kinase AKT [1][7][54]. As mentioned earlier, MSA exhibits angiogenesis inhibitory properties. In investigations on angiogenesis inhibition, downregulation of the expression and levels of hypoxia-inducible factor 1α (HIF-1a), vascular endothelial growth factor (VEGF), matrix metalloproteinase-2 (MMP-2), angiopoietin-2 (Ang-2) and integrin β3 (ITGB3, CD61) was observed [45][7][58][61]. In turn, disorganization of CD61 aggregates interrupted the phosphorylation of AKT, IκBα, and NF-κB [45]. Apart from apoptosis, there are also other types of cell death. Interestingly, it was observed that MSA induced necrosis in MCF-7 [13] and U-2 OS [18] cells.
In the course of the research, it was also found that MSA enhances the effectiveness of several chemotherapy drugs, i.e., paclitaxel [63][64], TAM [56], doxorubicin, cytosine arabinoside [2], taxol, etoposide, 7-ethyl-10-hydroxycamptotecin (SN-38) [63], and cyclophosphamide [65]. The applied combination of MSA (4.5 mg/kg b.w./day) and paclitaxel (microtubule inhibitor; 10 mg/k b.w./day) in the xenografts of triple-negative breast cancer in SCID mice showed synergistic enhancement of the therapeutic effect of paclitaxel, which was the result of arresting the cell cycle at the G2/M checkpoint and a significantly higher percentage of apoptotic cells in which the induction of apoptosis occurred through caspases [64]. In a study by Hu et al. [63], MSA amplified the apoptotic effects of paclitaxel (10 µM), etoposide (topoisomerase II inhibitor; 15 µM), and SN-38 (topoisomerase I inhibitor) in prostate cancer DU145 cells by affecting JNK-dependent molecular targets, amplifying the cascade of caspase 8 activity. Additionally, it was found that MSA can intensify cell uptake/retention of SN-38.
In turn, Li et al. [56] concluded that combination therapy of TAM + MSA against TAM-resistant and TAM-sensitive breast cancer cell lines increased the effectiveness of TAM and sensitization of TAM-resistant cells. The sensitization mechanism consisted of arresting the cell cycle in phase G1 by TAM, which permitted more cells to enter the mitochondrial apoptotic pathway induced by MSA. Another possible mechanism for MSA + TAM is a loss of ERα signalization. Among other drugs, MSA in combination with ABT-737 (inhibitor of Bcl-2 family proteins) increased caspase 8, 9, and 3 activity, introducing more cells into the apoptotic pathway in PC-3, MDA-MB-231, and HT-29 cell lines [3].
Additionally, contrary to selenite, MSA synergistically enhanced the proapoptotic properties of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) in DU145 prostate cancer cells, which was associated with the internal mitochondrial pathway [11]. Except for the synergistic enhancing efficacy of commonly used cytostatic agents by MSA, Lafin et al. [66] recently found that this compound also sensitizes head and neck squamous cell carcinoma (HNSCC) to radiotherapy. The mechanism involved in this phenomenon was the induction of lipid peroxidation (LPO) by MSA. Very importantly, MSA was non-toxic to normal skin cells. These results suggest that MSA may be an effective adjuvant during radiotherapy in HNSCC. In conclusion, MSA has very promising anticancer potential both alone and in combination with other cytotoxic drugs and radiation.
2.6. Selenoesters
The design and synthesis of alkyl and aryl selenoesters were guided by the creation of compounds that would be enzymatically reduced (may be TrxR1 substrates [67]) or hydrolyzed (mediated by a nucleophilic agent such as water [68] or enzymes such ascarbonic anhydrases [69] or acetylcholinesterase [70]) to metabolites (i.e., selenol, methylselenol) exhibiting redox activity in cells. These small active molecules can disrupt redox processes in cells, finally leading to cancer cell death [45]. The cytotoxicity of these derivatives has been studied on colon, prostate, breast, lung, and T-lymphoma cell lines, showing activity even at nanomolar concentrations [45][71][72]. Selenoesters with ketone end fragments showed the highest activity (Figure 7, structures 19–21). Moreover, derivatives with ketone terminal fragments inhibited the efflux pump ABCB1, responsible for MDR, more strongly than the reference compound verapamil. In mouse MDR T-lymphoma cells, the strongest inhibitor of ABCB1 (glycoprotein P, P-gp) was Se-2-oxopropyl 4-chlorobenzoselenoate (Figure 7, structure 19), which was 3.4-fold more potent than verapamil [72].
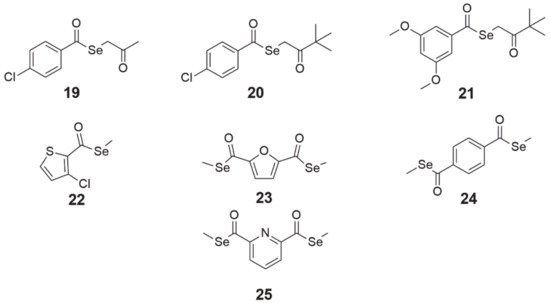
Figure 7. Chemical structures of selenoester derivatives (19—Se-2-oxopropyl 4-chlorobenzoselenoate, 20—3,3-dimethyl-2-oxobutyl 4-chlorobenzoselenoate, 21—3,3-dimethyl-2-oxobutyl 3,5-dimethoxybenzoselenoate, 22—methyl 3-chlorothiophen-2-carboselenoate, 23—dimethyl 2,5-furandicarboselenoate, 24—dimethyl benzene-1,4-dicarboselenoate, 25—dimethyl pyridine-2,6-dicarboselenoate).
Similarly, in the case of MDR Colo 320 human adenocarcinoma cells, the same compound 19 exhibited a four-fold greater inhibition of P-gp than the reference compound and was the most selective of the most active compounds 19–21 in this study [22]. It is worth noting that in both studies, the concentration of compound 19 was 10-fold lower than verapamil (2 µM 19, 20 µM verapamil) [22][72]. All compounds 19–21 induced apoptosis in mouse MDR T-lymphoma cells, wherein compounds 20 and 21 induced early and compound 19 induced late apoptosis [72]. In addition, these compounds 19–21 seem to be interesting agents for use as adjuvants in chemotherapy. In the experiments carried out by Spengler et al. [73], it was observed that compounds 19–21 exerted synergistic interactions with topotecan and vincristine, whereas additionally compounds 19 and 20 interacted with doxorubicin (Dox) and compound 19 interacted with cyclophosphamide. This would suggest that these compounds could affect the formation of microtubules and cell topoisomerases. However, it is surprising that these compounds 19–21 interacted antagonistically with verapamil, which, in comparison with the results of the two previous studies, where inhibition of efflux pump ABCB1 was shown, would suggest possible competition for a transporter between compounds 19–21 and verapamil.
Compounds 22 (methyl 3-chlorothiophen-2-carboselenoate, Figure 7) and 23 (dimethyl 2,5-furandicarboselenoate, Figure 7) were tested on cancer cell lines of the pancreas (PANC-1), lung (HTB-54), breast (MCF-7), prostate (PC-3), colorectal (HT-29), and chronic myeloid leukemia (K-562) [68][67]. Díaz-Argelich et al. [67] found that cell proliferation was inhibited in micromolar concentrations <10 µM (except for K-562 cells, where IC50 was 38.7 and 42 µM for 22 and 23, respectively) with cell cycle arrest in the G2/M phase. Growth inhibition was induced by these agents in a dose-dependent manner, whereas the effects of compound 23 were additionally time-dependent. The investigators’ assumption was the synthesis of compounds which, after hydrolysis, would release methylselenol, a key metabolite with anticancer properties. However, during the study, it turned out that compounds 22 and 23 are TrxR1 substrates and a reduction through this enzyme is more effective than hydrolysis, which could increase methylselenol generation. In addition, it was confirmed that they did not exhibit free radical scavenging properties and that cell death partly depended on the caspase pathway.
Entosis is a form of cancer cell cannibalism, in which a living cell is absorbed into the cytoplasm of another cell and then digested by lysosomal enzymes. This process is based on the principle of autophagy but without the presence of autophagosomes [74]. In another study carried out by Khalkar et al. [68], it was observed that derivatives of methylselenoesters 22 and 23 caused a drop in cell adhesion to the surface as a result of a decrease in the expression of cell division control protein 42 homolog (CD42) and a discontinuation of integrin β1 (CD29) signaling. The detachment of cells and a simultaneous increase in N-cadherin levels caused the cells to clump into grape-like aggregates, which led to the absorption of one cell by another (entosis) and finally the death of the trapped cell.
The compounds 24 (dimethyl benzene-1,4-dicarboselenoate, Figure 7) and 25 (dimethyl pyridine-2,6-dicarboselenoate, Figure 7), synthesized by Domínguez-Álvarez et al. [71], were tested for their cytotoxicity. Treatment with compounds 24 and 25 in nanomolar concentrations inhibited the growth of PC-3 prostate cancer cells. Furthermore, derivatives 24 and 25 were the most selective among all the compounds in the panel, as well as the reference drugs (etoposide, cisplatin), and did not exhibit antioxidant properties, the same as 19. In addition, both compounds interacted synergistically with vincristine, cyclophosphamide, Dox, and methotrexate (MTX) in concentrations higher than 19–21 and showed moderate synergy with verapamil [73]. Regarding their effect on MDR, 24 and 25 marginally affected the activity of the P-gp efflux pump; their effect at both concentrations (2 and 20 µM) was lower than that of verapamil in both mouse MDR T-lymphoma cells [72] and MDR Colo 320 human adenocarcinoma cells [22].
2.7. Selol
Selol is a very interesting compound containing in its structure selenium at +4 oxidation stage [75]. It was obtained at the Medical University of Warsaw as a result of a reaction of triglycerides from sunflower oil with selenic acid (IV) [76][77]. The study, conducted by Rahden-Staroń [75], showed that this compound is non-toxic and non-mutagenic. However, its toxicity to the body depends on the route of administration. Selol administered parenterally was non-toxic, whereas during oral administration its toxicity increased sharply, which may indicate the formation of more harmful products during digestion [76]. For this reason, it should be considered to be administered only parenterally. Moreover, the activity of Selol is determined by its Se content—the number of dioxaselenolane rings (Selol 2%—single rings, 5%> single and double rings, Figure 8). The authors found that 2% Selol exerted antioxidant activity through the induction of enzymes of the second phase of detoxification, which would prove its chemopreventive properties, while 7% Selol exhibited cytotoxic properties on human colorectal adenocarcinoma Caco-2 cells [78].
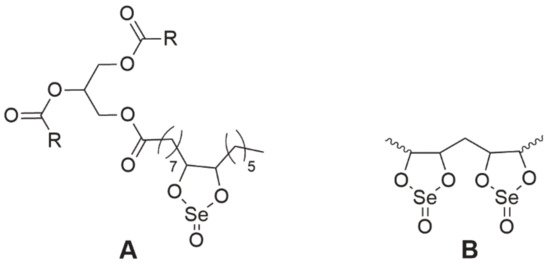
Figure 8. The chemical structure of Selol. A—2% Selol (single dioxaselenolane rings), B—5% Selol, and more (single and double dioxaselenolane rings). R, polyunsaturated fatty acid moiety.
Additionally, it is worth noting that its antioxidant activity and high bioavailability result from the incorporation of Se from Selol into selenoproteins [79]. Selol was tested on various cancer cell lines, including prostate cancer and leukemia [80][81]. In vitro experiments on HL-60 and vicristine/doxorubicin-resistant (HL-60/Vinc and HL-60/Dox) leukemia cells showed that Selol inhibited cell proliferation and induced apoptosis, affecting resistant lines more strongly [80]. In the androgen-dependent prostate cancer (LNCaP) cell line, this compound inhibited cell proliferation and probably directed them to the apoptotic pathway, not showing this activity against normal cells [81]. Additionally, 5% Selol is a prooxidant (causes excessive ROS production) [82] and was also recognized as a TrxR inhibitor by Sochacka et al. [83].
In research on the interactions of this compound with the commonly available drugs used in chemotherapy, it was reported that it enhanced the antiproliferative effects of Dox, especially in cells that were resistant to this drug [84]. Most importantly, in vincristine-induced hyperalgesia, Selol enhanced the analgesic effects of fentanyl, buprenorphine, and morphine, which gives hope for a new therapy in palliative care in terminal states of cancers [85]. Despite the promising results, the potential molecular targets and the detailed mechanism of Selol’s activity need to be thoroughly investigated in the future.
References
- Sinha, R.; El-Bayoumy, K. Apoptosis is a critical cellular event in cancer chemoprevention and chemotherapy by selenium compounds. Curr. Cancer Drug Targets 2004, 4, 13–28.
- Tan, H.W.; Mo, H.-Y.; Lau, A.T.Y.; Xu, Y.-M. Selenium species: Current status and potentials in cancer prevention and therapy. Int. J. Mol. Sci. 2018, 20, 75.
- Sanmartín, C.; Plano, D.; Sharma, A.K.; Palop, J.A. Selenium compounds, apoptosis and other types of cell death: An overview for cancer therapy. Int. J. Mol. Sci. 2012, 13, 9649–9672.
- Naithani, R. Organoselenium compounds in cancer chemoprevention. Mini Rev. Med. Chem. 2008, 8, 657–668.
- Li, G.-X.; Hu, H.; Jiang, C.; Schuster, T.; Lü, J. Differential involvement of reactive oxygen species in apoptosis induced by two classes of selenium compounds in human prostate cancer cells. Int. J. Cancer 2007, 120, 2034–2043.
- Gasparian, A.V.; Yao, Y.J.; Lü, J.; Yemelyanov, A.Y.; Lyakh, L.A.; Slaga, T.J.; Budunova, I.V. Selenium compounds inhibit IκB Kinase (IKK) and Nuclear Factor-κB (NF-κB) in prostate cancer cells. Mol. Cancer Ther. 2002, 1, 1079–1087.
- Jiang, C.; Wang, Z.; Ganther, H.; Lü, J. Distinct effects of methylseleninic acid versus selenite on apoptosis, cell cycle, and protein kinase pathways in DU145 human prostate cancer cells. Mol. Cancer Ther. 2002, 1, 1059–1066.
- Li, G.-x.; Lee, H.-J.; Wang, Z.; Hu, H.; Liao, J.D.; Watts, J.C.; Combs, G.F., Jr.; Lü, J. Superior in vivo inhibitory efficacy of methylseleninic acid against human prostate cancer over selenomethionine or selenite. Carcinogenesis 2008, 29, 1005–1012.
- Shen, C.L.; Song, W.; Pence, B.C. Interactions of selenium compounds with other antioxidants in DNA damage and apoptosis in human normal keratinocytes. Cancer Epidemiol. Biomarkers Prev. 2001, 10, 385–390.
- Gopalakrishna, R.; Gundimeda, U. Protein kinase C as a molecular target for cancer prevention by selenocompounds. Nutr. Cancer 2001, 40, 55–63.
- Rikiishi, H. Apoptotic cellular events for selenium compounds involved in cancer prevention. J. Bioenerg. Biomembr. 2007, 39, 91–98.
- Zhou, N.; Xiao, H.; Li, T.K.; Nur, E.K.A.; Liu, L.F. DNA damage-mediated apoptosis induced by selenium compounds. J. Biol. Chem. 2003, 278, 29532–29537.
- De Miranda, J.X.; Andrade, F.d.O.; Conti, A.d.; Dagli, M.L.Z.; Moreno, F.S.; Ong, T.P. Effects of selenium compounds on proliferation and epigenetic marks of breast cancer cells. J. Trace Elem. Med. Biol. 2014, 28, 486–491.
- Liu, C.; Chen, J.; Wu, Z.; Li, Y.; Zhu, Y.; Ren, Y.; Zhou, Q. Selenium compounds induce ROS in human high-metastatic large cell lung cancer cell line L9981. Zhongguo Fei Ai Za Zhi 2008, 11, 354–358.
- Shen, H.-M.; Ding, W.-X.; Ong, C.-N. Intracellular glutathione is a cofactor in methylseleninic acid-induced apoptotic cell death of human hepatoma HEPG2 cells. Free Radic. Biol. Med. 2002, 33, 552–561.
- Soukupová, K.; Rudolf, E. Suppression of proliferation and activation of cell death by sodium selenite involves mitochondria and lysosomes in chemoresistant bladder cancer cells. J. Trace Elem. Med. Biol. 2019, 52, 58–67.
- Harmanci, D.; Erbayraktar, Z.; Sayin, O.; Guner, G.A. In vitro effects of selenium on human glioblastoma multiforme cell lines: A preliminary study. Acta Clin. Croat. 2017, 56, 48–57.
- Pang, K.-L.; Chin, K.-Y. Emerging Anticancer Potentials of Selenium on Osteosarcoma. Int. J. Mol. Sci. 2019, 20, 5318.
- Shigemi, Z.; Manabe, K.; Hara, N.; Baba, Y.; Hosokawa, K.; Kagawa, H.; Watanabe, T.; Fujimuro, M. Methylseleninic acid and sodium selenite induce severe ER stress and subsequent apoptosis through UPR activation in PEL cells. Chem. Biol. Interact. 2017, 266, 28–37.
- Kim, T.-S.; Yun, B.Y.; Kim, I.Y. Induction of the mitochondrial permeability transition by selenium compounds mediated by oxidation of the protein thiol groups and generation of the superoxide. Biochem. Pharmacol. 2003, 66, 2301–2311.
- Subburayan, K.; Thayyullathil, F.; Pallichankandy, S.; Cheratta, A.R.; Galadari, S. Superoxide-mediated ferroptosis in human cancer cells induced by sodium selenite. Transl. Oncol. 2020, 13, 100843.
- Gajdács, M.; Spengler, G.; Sanmartín, C.; Marć, M.A.; Handzlik, J.; Domínguez-Álvarez, E. Selenoesters and selenoanhydrides as novel multidrug resistance reversing agents: A confirmation study in a colon cancer MDR cell line. Bioorg. Med. Chem. Lett. 2017, 27, 797–802.
- ClinicalTrials.gov. Study Evaluating Sodium Selenite in Combination with Abiraterone in Castrate Resistant Prostate Cancer Progressing on Abiraterone. Available online: (accessed on 7 January 2021).
- ClinicalTrials.gov. Phase I Sodium Selenite in Combination with Docetaxel in Castration-resistant Prostate Cancer. Available online: (accessed on 7 January 2021).
- ClinicalTrials.gov. Sodium Selenite and Radiation Therapy in Treating Patients with Metastatic Cancer. Available online: (accessed on 7 January 2021).
- ClinicalTrials.gov. The Use of Selenium to Treat Secondary Lymphedema—Breast Cancer. Available online: (accessed on 7 January 2021).
- ClinicalTrials.gov. Sodium Selenite as a Cytotoxic Agent in Advanced Carcinoma (SECAR). Available online: (accessed on 7 January 2021).
- ClinicalTrials.gov. High Dose Inorganic Selenium for Preventing Chemotherapy Induced Peripheral Neuropathy (SELENIUM). Available online: (accessed on 7 January 2021).
- Álvarez-Pérez, M.; Ali, W.; Marć, M.A.; Handzlik, J.; Domínguez-Álvarez, E. Selenides and Diselenides: A Review of Their Anticancer and Chemopreventive Activity. Molecules 2018, 23, 628.
- Brigelius-Flohé, R. Selenium compounds and selenoproteins in cancer. Chem. Biodivers. 2008, 5, 389–395.
- Nedel, F.; Campos, V.F.; Alves, D.; McBride, A.J.A.; Dellagostin, O.A.; Collares, T.; Savegnago, L.; Seixas, F.K. Substituted diaryl diselenides: Cytotoxic and apoptotic effect in human colon adenocarcinoma cells. Life Sci. 2012, 91, 345–352.
- Posser, T.; de Paula, M.T.; Franco, J.L.; Leal, R.B.; da Rocha, J.B.T. Diphenyl diselenide induces apoptotic cell death and modulates ERK1/2 phosphorylation in human neuroblastoma SH-SY5Y cells. Arch. Toxicol. 2011, 85, 645–651.
- Kim, C.; Lee, J.; Park, M.-S. Synthesis of new diorganodiselenides from organic halides: Their antiproliferative effects against human breast cancer MCF-7 cells. Arch. Pharmacal. Res. 2015, 38, 659–665.
- Krasowska, D.; Iraci, N.; Santi, C.; Drabowicz, J.; Cieslak, M.; Kaźmierczak-Barańska, J.; Palomba, M.; Królewska-Golińska, K.; Magiera, J.; Sancineto, L. Diselenides and benzisoselenazolones as antiproliferative agents and glutathione-s-transferase inhibitors. Molecules 2019, 24, 2914.
- Nogueira, C.W.; Rocha, J.B. Toxicology and pharmacology of selenium: Emphasis on synthetic organoselenium compounds. Arch. Toxicol. 2011, 85, 1313–1359.
- Rosa, R.M.; Roesler, R.; Braga, A.L.; Saffi, J.; Henriques, J.A.P. Pharmacology and toxicology of diphenyl diselenide in several biological models. Braz. J. Med. Biol. Res. 2007, 40, 1287–1304.
- Meinerz, D.F.; Allebrandt, J.; Mariano, D.O.C.; Waczuk, E.P.; Soares, F.A.; Hassan, W.; Rocha, J.B.T. Differential genotoxicity of diphenyl diselenide (PhSe)2 and diphenyl ditelluride (PhTe)2. PeerJ 2014, 2, e290.
- Stefanello, S.T.; Dobrachinski, F.; de Carvalho, N.R.; Amaral, G.P.; Barcelos, R.P.; Oliveira, V.A.; Oliveira, C.S.; Giordani, C.F.A.; Pereira, M.E.; Rodrigues, O.E.D.; et al. Free radical scavenging in vitro and biological activity of diphenyl diselenide-loaded nanocapsules: DPDS-NCS antioxidant and toxicological effects. Int. J. Nanomed. 2015, 10, 5663–5670.
- Chagas, P.M.; Bortolatto, C.F.; Wilhelm, E.A.; Nogueira, C.W. High doses of 2,2′-dithienyl diselenide cause systemic toxicity in rats: An in vitro and in vivo study. J. Appl. Toxicol. 2013, 33, 480–487.
- Savegnago, L.; Jesse, C.R.; Nogueira, C.W. Structural modifications into diphenyl diselenide molecule do not cause toxicity in mice. Environ. Toxicol. Pharmacol. 2009, 27, 271–276.
- Marcondes Sari, M.H.; Ferreira, L.M.; Zborowski, V.A.; Araujo, P.C.O.; Cervi, V.F.; Brüning, C.A.; Cruz, L.; Nogueira, C.W. p,p’-Methoxyl-diphenyl diselenide-loaded polymeric nanocapsules are chemically stable and do not induce toxicity in mice. Eur. J. Pharm. Biopharm. 2017, 117, 39–48.
- Dos Santos, E.d.A.; Hamel, E.; Bai, R.; Burnett, J.C.; Tozatti, C.S.S.; Bogo, D.; Perdomo, R.T.; Antunes, A.M.M.; Marques, M.M.; Matos, M.d.F.C.; et al. Synthesis and evaluation of diaryl sulfides and diaryl selenide compounds for antitubulin and cytotoxic activity. Bioorg. Med. Chem. Lett. 2013, 23, 4669–4673.
- Zhang, L.; Zhou, L.; Du, J.; Li, M.; Qian, C.; Cheng, Y.; Peng, Y.; Xie, J.; Wang, D. Induction of apoptosis in human multiple myeloma cell lines by ebselen via enhancing the endogenous reactive oxygen species production. Biomed Res. Int. 2014, 2014, 696107.
- Yang, C.F.; Shen, H.M.; Ong, C.N. Protective effect of ebselen against hydrogen peroxide-induced cytotoxicity and DNA damage in HepG2 cells. Biochem. Pharmacol. 1999, 57, 273–279.
- Gandin, V.; Khalkar, P.; Braude, J.; Fernandes, A.P. Organic selenium compounds as potential chemotherapeutic agents for improved cancer treatment. Free Radic. Biol. Med. 2018, 127, 80–97.
- Chen, Z.; Lai, H.; Hou, L.; Chen, T. Rational design and action mechanisms of chemically innovative organoselenium in cancer therapy. Chem. Commun. 2020, 56, 179–196.
- Sharma, V.; Tewari, R.; Sk, U.H.; Joseph, C.; Sen, E. Ebselen sensitizes glioblastoma cells to Tumor Necrosis Factor (TNFalpha)-induced apoptosis through two distinct pathways involving NF-kappaB downregulation and Fas-mediated formation of death inducing signaling complex. Int. J. Cancer 2008, 123, 2204–2212.
- Thabet, N.M.; Moustafa, E.M. Synergistic effect of Ebselen and gamma radiation on breast cancer cells. Int. J. Radiat. Biol. 2017, 93, 784–792.
- ClinicalTrials.gov. Study to Evaluate the Safety and Pharmacokinetics of SPI-1005. Available online: (accessed on 7 January 2021).
- ClinicalTrials.gov. SPI-1005 for Prevention and Treatment of Chemotherapy Induced Hearing Loss. Available online: (accessed on 7 January 2021).
- Wang, L.; Yang, Z.; Fu, J.; Yin, H.; Xiong, K.; Tan, Q.; Jin, H.; Li, J.; Wang, T.; Tang, W.; et al. Ethaselen: A potent mammalian thioredoxin reductase 1 inhibitor and novel organoselenium anticancer agent. Free Radic. Biol. Med. 2012, 52, 898–908.
- Tan, Q.; Li, J.; Yin, H.-w.; Wang, L.-h.; Tang, W.-c.; Zhao, F.; Liu, X.-m.; Zeng, H.-h. Augmented antitumor effects of combination therapy of cisplatin with ethaselen as a novel thioredoxin reductase inhibitor on human A549 cell in vivo. Investig. New Drugs 2010, 28, 205–215.
- ClinicalTrials.gov. Ethaselen for the Treatment of Thioredoxin Reductase High Expression Advanced Non-small Cell Lung Cancers. Available online: (accessed on 27 November 2020).
- Sinha, I.; Allen, J.E.; Pinto, J.T.; Sinha, R. Methylseleninic acid elevates REDD1 and inhibits prostate cancer cell growth despite AKT activation and mTOR dysregulation in hypoxia. Cancer Med. 2014, 3, 252–264.
- Zeng, H.; Wu, M. The inhibitory efficacy of methylseleninic acid against colon cancer xenografts in C57BL/6 Mice. Nutr. Cancer 2015, 67, 831–838.
- Li, Z.; Carrier, L.; Rowan, B.G. Methylseleninic acid synergizes with tamoxifen to induce caspase-mediated apoptosis in breast cancer cells. Mol. Cancer Ther. 2008, 7, 3056–3063.
- Wang, L.; Bonorden, M.J.L.; Li, G.-x.; Lee, H.-J.; Hu, H.; Zhang, Y.; Liao, J.D.; Cleary, M.P.; Lü, J. Methyl-selenium compounds inhibit prostate carcinogenesis in the transgenic adenocarcinoma of mouse prostate model with survival benefit. Cancer Prev. Res. 2009, 2, 484–495.
- Sinha, I.; Null, K.; Wolter, W.; Suckow, M.A.; King, T.; Pinto, J.T.; Sinha, R. Methylseleninic acid downregulates hypoxia-inducible factor-1α in invasive prostate cancer. Int. J. Cancer 2012, 130, 1430–1439.
- Gundimeda, U.; Schiffman, J.E.; Chhabra, D.; Wong, J.; Wu, A.; Gopalakrishna, R. Locally generated methylseleninic acid induces specific inactivation of protein kinase C isoenzymes: Relevance to selenium-induced apoptosis in prostate cancer cells. J. Biol. Chem. 2008, 283, 34519–34531.
- Cho, S.D.; Jiang, C.; Malewicz, B.; Dong, Y.; Young, C.Y.F.; Kang, K.-S.; Lee, Y.-S.; Ip, C.; Lü, J. Methyl selenium metabolites decrease prostate-specific antigen expression by inducing protein degradation and suppressing androgen-stimulated transcription. Mol. Cancer Ther. 2004, 3, 605–612.
- Wu, X.; Zhang, Y.; Pei, Z.; Chen, S.; Yang, X.; Chen, Y.; Lin, D.; Ma, R.Z. Methylseleninic acid restricts tumor growth in nude mice model of metastatic breast cancer probably via inhibiting angiopoietin-2. BMC Cancer 2012, 12, 192.
- Wang, L.; Walia, B.; Evans, J.; Gewirtz, A.T.; Merlin, D.; Sitaraman, S.V. IL-6 Induces NF-κB Activation in the Intestinal Epithelia. J. Immunol. 2003, 171, 3194–3201.
- Hu, H.; Jiang, C.; Ip, C.; Rustum, Y.M.; Lü, J. Methylseleninic Acid Potentiates Apoptosis Induced by Chemotherapeutic Drugs in Androgen-Independent Prostate Cancer Cells. Clin. Cancer Res. 2005, 11, 2379–2388.
- Qi, Y.; Fu, X.; Xiong, Z.; Zhang, H.; Hill, S.M.; Rowan, B.G.; Dong, Y. Methylseleninic acid enhances paclitaxel efficacy for the treatment of triple-negative breast cancer. PLoS ONE 2012, 7, e31539.
- Liu, Y.; Li, W.; Guo, M.; Li, C.; Qiu, C. Protective Role of Selenium Compounds on the Proliferation, Apoptosis, and Angiogenesis of a Canine Breast Cancer Cell Line. Biol. Trace Elem. Res. 2016, 169, 86–93.
- Lafin, J.T.; Sarsour, E.H.; Kalen, A.L.; Wagner, B.A.; Buettner, G.R.; Goswami, P.C. Methylseleninic acid induces lipid peroxidation and radiation sensitivity in head and neck cancer cells. Int. J. Mol. Sci. 2019, 20, 225.
- Díaz-Argelich, N.; Encío, I.; Plano, D.; Fernandes, A.P.; Palop, J.A.; Sanmartín, C. Novel methylselenoesters as antiproliferative agents. Molecules 2017, 22, 1288.
- Khalkar, P.; Díaz-Argelich, N.; Antonio Palop, J.; Sanmartín, C.; Fernandes, A.P. Novel methylselenoesters induce programed cell death via entosis in pancreatic cancer cells. Int. J. Mol. Sci. 2018, 19, 2849.
- Angeli, A.; Carta, F.; Supuran, C.T. Carbonic anhydrases: Versatile and useful biocatalysts in chemistry and biochemistry. Catalysts 2020, 10, 1008.
- Delfino, R.T.; Ribeiro, T.S.; Figueroa-Villar, J.D. Organophosphorus compounds as chemical warfare agents: A review. J. Braz. Chem. Soc. 2009, 20, 407–428.
- Domínguez-Álvarez, E.; Plano, D.; Font, M.; Calvo, A.; Prior, C.; Jacob, C.; Palop, J.A.; Sanmartín, C. Synthesis and antiproliferative activity of novel selenoester derivatives. Eur. J. Med. Chem. 2014, 73, 153–166.
- Domínguez-Álvarez, E.; Gajdács, M.; Spengler, G.; Palop, J.A.; Marć, M.A.; Kieć-Kononowicz, K.; Amaral, L.; Molnár, J.; Jacob, C.; Handzlik, J.; et al. Identification of selenocompounds with promising properties to reverse cancer multidrug resistance. Bioorg. Med. Chem. Lett. 2016, 26, 2821–2824.
- Spengler, G.; Gajdács, M.; Marć, M.A.; Domínguez-Álvarez, E.; Sanmartín, C. Organoselenium compounds as novel adjuvants of chemotherapy drugs-a promising approach to fight cancer drug resistance. Molecules 2019, 24, 336.
- White, E. Entosis: It’s a cell-eat-cell world. Cell 2007, 131, 840–842.
- Rahden-Staroń, I.; Suchocki, P.; Czeczot, H. Evaluation of mutagenic activity of the organo-selenium compound Selol by use of the Salmonella typhimurium mutagenicity assay. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2010, 699, 44–46.
- Jastrzebski, Z.; Czyzewska-Szafran, H.; Fijatek, Z.; Suchocki, P.; Fitak, B.A. Toxicity studies of a new selenium compound, Selol, in rats. Drugs Exp. Clin. Res. 1995, 21, 217–220.
- Ślusarczyk, J.; Dudek, M.; Wierzbicka, M.; Suchocki, P.; Kuraś, M. Antimitotic effect of Selol and sodium selenate (IV) on Allium test cells. Caryologia 2014, 67, 250–259.
- Suchocki, P.; Misiewicz-Krzemińska, I.; Skupińska, K.; Niedźwiecka, K.; Lubelska, K.; Fijałek, Z.; Kasprzycka-Guttman, T. Selenitetriglicerydes affect CYP1A1 and QR activity by involvement of reactive oxygen species and Nrf2 transcription factor. Pharmacol. Rep. 2010, 62, 352–361.
- Sonet, J.; Mosca, M.; Bierla, K.; Modzelewska, K.; Flis-Borsuk, A.; Suchocki, P.; Ksiazek, I.; Anuszewska, E.; Bulteau, A.-L.; Szpunar, J.; et al. Selenized plant oil is an efficient source of selenium for selenoprotein biosynthesis in human cell lines. Nutrients 2019, 11, 1524.
- Suchocki, P.; Misiewicz, I.; Skupinska, K.; Waclawek, K.; Fijalek, Z.; Kasprzycka-Guttman, T. The activity of Selol in multidrug-resistant and sensitive human leukemia cells. Oncol. Rep. 2007, 18, 893–899.
- Flis, A.; Suchocki, P.; Królikowska, M.A.; Suchocka, Z.; Remiszewska, M.; Śliwka, L.; Książek, I.; Sitarz, K.; Sochacka, M.; Hoser, G.; et al. Selenitetriglycerides-Redox-active agents. Pharmacol. Rep. 2015, 67, 1–8.
- Grosicka-Maciąg, E.; Kurpios-Piec, D.; Woźniak, K.; Kowalewski, C.; Szumiło, M.; Drela, N.; Kiernozek, E.; Suchocki, P.; Rahden-Staroń, I. Selol (Se IV) modulates adhesive molecules in control and TNF-α-stimulated HMEC-1 cells. J. Trace Elem. Med. Biol. 2019, 51, 106–114.
- Sochacka, M.; Giebułtowicz, J.; Remiszewska, M.; Suchocki, P.; Wroczyński, P. Effects of Selol 5% supplementation on the activity or concentration of antioxidants and malondialdehyde level in the blood of healthy mice. Pharmacol. Rep. 2014, 66, 301–310.
- Dudkiewicz-Wilczyńska, J.; Grabowska, A.; Książek, I.; Sitarz, K.; Suchocki, P.; Anuszewska, E. Comparison of selected gene expression profiles in sensitive and resistant cancer cells treated with doxorubicin and Selol. Contemp. Oncol. 2014, 18, 90–94.
- Bujalska, M.; Gumułka, S.W. Effect of selenium compound (selol) on the opioid activity in vincristine induced hyperalgesia. Neuro Endocrinol. Lett. 2008, 29, 552–557.