Netrin-1 is one of the most well-studied proteins that regulate axonal guidance and synaptogenesis via interaction with its receptors deleted in colorectal cancer (DCC) and UNC5H (unco-ordinated-5 homolog) to activate cell survival, differentiation and proliferation.
- intracerebral hemorrhage
- hemin
- neuroprotection
- Netrin-1
- peptide
1. Introduction
Intracerebral hemorrhage (ICH) is a devastating stroke subtype with a high mortality and morbidity rate [1][2][3] that involves abnormal rupture of cerebral blood vessels within the brain [4][5]. Surgical removal of hematomas is preferred for achieving hemostasis and relieving intracranial pressure [6]. However, remnant hematic residues and blood breakdown products can still promote secondary brain injury following ICH, inducing neuronal damage and delaying functional recovery [7][8][9]. Thus, the development of effective strategies targeting secondary brain injury induced neuronal damage would be beneficial for stroke treatment.
Intracerebral hemorrhage (ICH) is a devastating stroke subtype with a high mortality and morbidity rate [1,2,3] that involves abnormal rupture of cerebral blood vessels within the brain [4,5]. Surgical removal of hematomas is preferred for achieving hemostasis and relieving intracranial pressure [6]. However, remnant hematic residues and blood breakdown products can still promote secondary brain injury following ICH, inducing neuronal damage and delaying functional recovery [7,8,9]. Thus, the development of effective strategies targeting secondary brain injury induced neuronal damage would be beneficial for stroke treatment.
Netrin-1 is one of the most well-studied proteins that regulate axonal guidance and synaptogenesis [10][11][12] via interaction with its receptors deleted in colorectal cancer (DCC) and UNC5H (unco-ordinated-5 homolog) to activate cell survival, differentiation and proliferation [13][14][15]. The receptors induce a death signal to mediate apoptosis when the Netrin-1 is absent [16]. It is known that the binding of Netrin-1 with DCC results in dimerization of DCC and initiates tyrosine 861 phosphorylation of focal adhesion kinase (FAK) to induce neurite outgrowth and axonal guidance [17][18]. The binding of Netrin-1 with DCC involves the EGF3 domain of Netrin-1 and the FN5 domain of DCC in activation of Netrin-1 signaling pathway [19][20]. Recently, Netrin-1 was shown to exert neuroprotective effects in ischemic regions [21]. The expression level of Netrin-1, which inhibits cell apoptosis and promotes neuronal regeneration, in the serum is positively correlated with patient recovery from ischemic stroke [22][23].
Netrin-1 is one of the most well-studied proteins that regulate axonal guidance and synaptogenesis [10,11,12] via interaction with its receptors deleted in colorectal cancer (DCC) and UNC5H (unco-ordinated-5 homolog) to activate cell survival, differentiation and proliferation [13,14,15]. The receptors induce a death signal to mediate apoptosis when the Netrin-1 is absent [16]. It is known that the binding of Netrin-1 with DCC results in dimerization of DCC and initiates tyrosine 861 phosphorylation of focal adhesion kinase (FAK) to induce neurite outgrowth and axonal guidance [17,18]. The binding of Netrin-1 with DCC involves the EGF3 domain of Netrin-1 and the FN5 domain of DCC in activation of Netrin-1 signaling pathway [19,20]. Recently, Netrin-1 was shown to exert neuroprotective effects in ischemic regions [21]. The expression level of Netrin-1, which inhibits cell apoptosis and promotes neuronal regeneration, in the serum is positively correlated with patient recovery from ischemic stroke [22,23].
Interestingly, transplantation of bone marrow mesenchymal stem cells that secrete Netrin-1 into the damaged rat sciatic nerve initiates axonal regeneration [24] and reduces motor neuron death [25]. Stereotaxic injection of the Netrin-1 protein into the ventricles reduces nerve cell death after subarachnoid hemorrhage through activation of DCC/APPL-1/AKT signaling [26]. A polypeptide derived from the EGF3 domain of Netrin-1 (amino acids 423–433) markedly activates ERK phosphorylation and promotes the production of NO, which has cardioprotective effects [27][28]. However, Netrin-1 has been identified as a potential biomarker for tumorigenesis [29][30][31] because regulation of the Netrin-1 status is found in multiple tissue-derived cancers [32]. When Netrin-1 is bound to its receptor DCC or Unc5H, the receptors transduce a positive signal leading to survival, inflammation, angiogenesis and anti-apoptosis, which in turn regulates tumorigenesis [31]. Therefore, it is very important to identify the minimal functional domain of Netrin-1 that can preserve the neuroprotection and avoid the major side effects of Netrin-1.
Interestingly, transplantation of bone marrow mesenchymal stem cells that secrete Netrin-1 into the damaged rat sciatic nerve initiates axonal regeneration [24] and reduces motor neuron death [25]. Stereotaxic injection of the Netrin-1 protein into the ventricles reduces nerve cell death after subarachnoid hemorrhage through activation of DCC/APPL-1/AKT signaling [26]. A polypeptide derived from the EGF3 domain of Netrin-1 (amino acids 423–433) markedly activates ERK phosphorylation and promotes the production of NO, which has cardioprotective effects [27,28]. However, Netrin-1 has been identified as a potential biomarker for tumorigenesis [29,30,31] because regulation of the Netrin-1 status is found in multiple tissue-derived cancers [32]. When Netrin-1 is bound to its receptor DCC or Unc5H, the receptors transduce a positive signal leading to survival, inflammation, angiogenesis and anti-apoptosis, which in turn regulates tumorigenesis [31]. Therefore, it is very important to identify the minimal functional domain of Netrin-1 that can preserve the neuroprotection and avoid the major side effects of Netrin-1.
Here, we found that peptide E1 (residues 407–422), which is derived from the EGF3 domain of Netrin-1, interacts with DCC to protect neurons from hemin-induced toxicity. In an experimental model of ICH mice, the application of peptide E1 improved functional recovery, as determined by behavioral assays. Furthermore, peptide E1 can prevent neurons from degenerating after ICH. On the basis of these results, we identified a functional sequence of Netrin-1 that is involved in recovery after ICH and determined that this peptide may be used for the treatment of ICH.
2. EGF3 Domain Is Critical for the Netrin-DCC Interaction
Netrin-1 consists of a VI domain, three EGF repeats, and aC
-terminal domain [33]. The crystal structure of the human Netrin-1/DCC complex reveals that Netrin-1 EGF3 domain is crucial for the Netrin-1/DCC interaction, but not the EGF1 and EGF2 domains [34]. Mutagenesis key amino acid residues (His407, Gln442 or Gln443) at EGF3 domain completely disrupted Netrin-1/DCC binding [19]. EGF1/2 domains are required for switching attractive signaling into repulsive signaling when Unc5 coexists with DCC [20]. Thus, we first generated full-length and EGF3-deleted (Netrin-1 Δ407–443) Netrin-1 constructs (A) and transiently expressed these constructs in cultured HEK293T cells for GST pulldown. Binding was observed for the constructs that expressed Netrin-1 but not Netrin-1 (Δ407–443) (C). These results suggest that the domain of EGF3 containing amino acids 407–443 is responsible for the interaction of Netrin-1 with DCC.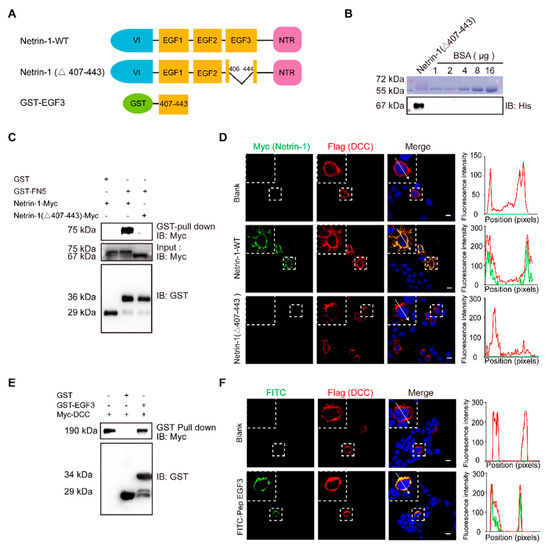
Figure 1.
A
B
C
D
E
F
3. The EGF3 Domain Induces Phosphorylation of Downstream Pathways of Netrin-1
The interaction of Netrin-1 and DCC activates multiple signaling pathways, including FAK, SFK and ERK [16][35]. Focal adhesion kinase (FAK) is one of the major tyrosine phosphorylation activities linked to Netrin-1/DCC signaling. Netrin-1 activates FAK phosphorylation and Src family protein tyrosine kinases (SFKs) phosphorylation [17]. Both FAK and SFK signaling pathways play a pivotal role in the developmental stage of nervous system and injury repair [36][37]. Moreover, Netrin-1 is able to activate the extracellular signal-regulated kinase-1/2 (ERK1/2), which mediates action of Mitogen-activated protein kinase (MAPK) pathway [38][39][40]. To investigate whether the EGF3 domain is critical for the activation of downstream signaling pathways via the binding of Netrin-1 to DCC, we first examined the effect of the EGF3 domain on the tyrosine 861 phosphorylation of FAK (
The interaction of Netrin-1 and DCC activates multiple signaling pathways, including FAK, SFK and ERK [16,35]. Focal adhesion kinase (FAK) is one of the major tyrosine phosphorylation activities linked to Netrin-1/DCC signaling. Netrin-1 activates FAK phosphorylation and Src family protein tyrosine kinases (SFKs) phosphorylation [17]. Both FAK and SFK signaling pathways play a pivotal role in the developmental stage of nervous system and injury repair [36,37]. Moreover, Netrin-1 is able to activate the extracellular signal-regulated kinase-1/2 (ERK1/2), which mediates action of Mitogen-activated protein kinase (MAPK) pathway [38,39,40]. To investigate whether the EGF3 domain is critical for the activation of downstream signaling pathways via the binding of Netrin-1 to DCC, we first examined the effect of the EGF3 domain on the tyrosine 861 phosphorylation of FAK (A). We found that deletion of EGF3 inhibited FAK phosphorylation in cultured cortical neurons (B,C), thus confirming an essential role for EGF3 in the Netrin-1 signaling pathway. Next, cultured cortical neurons were stimulated with the GST-EGF3 fusion protein for 20 min, and western blotting further confirmed that treatment with the GST-EGF3 fusion protein induced FAK, ERK and SFK phosphorylation (D–G). Taken together, these results suggest that the EGF3 domain of Netrin-1 functions as a potent stimulator to activate downstream signaling pathways.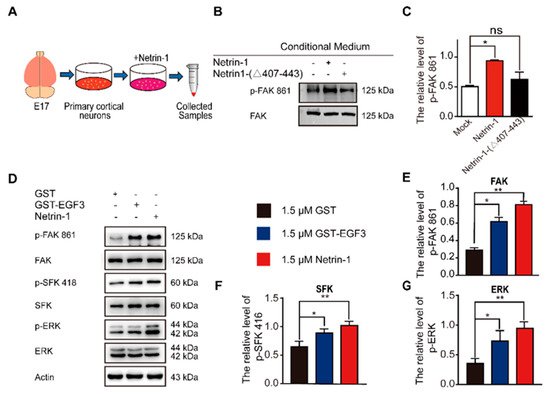
Figure 2.
A
B
C
B
n
D
E
G
D
n
p
p
4. Peptide E1 and E2 Interact with DCC to Activate the Downstream Signaling Pathway of Netrin-1
As previously described, a small peptide derived from the EGF3 domain of Netrin-1 (amino acids 423–433) induced ERK phosphorylation [27]. We synthesized peptides E1 (residues 407–422) and E2 (residues 423–433) that were derived from the EGF3 domain of Netrin-1 (
A). We found that incubation of peptide E1 increased the levels of phosphorylated FAK and SFK (
B,D,E) in the culture cells in a dose-dependent manner but did not alter the levels of phosphorylated ERK (
B,F). Unlike peptide E1, peptide E2 not only induced the tyrosine phosphorylation of FAK (
C,G) and the tyrosine phosphorylation of SFK (
C,H) but also induced the tyrosine phosphorylation of ERK (
C,I) in cultured cortical neurons. The control peptide (Pep Ctrl) had no detectable effect on the tyrosine phosphorylation of FAK, SFK and ERK (
). Furthermore, we observed that the phosphorylation of components of the FAK signaling pathway induced by peptide E1 was time-dependent (
). These results indicate that E1 and E2 are the minimal functional peptides of Netrin-1.
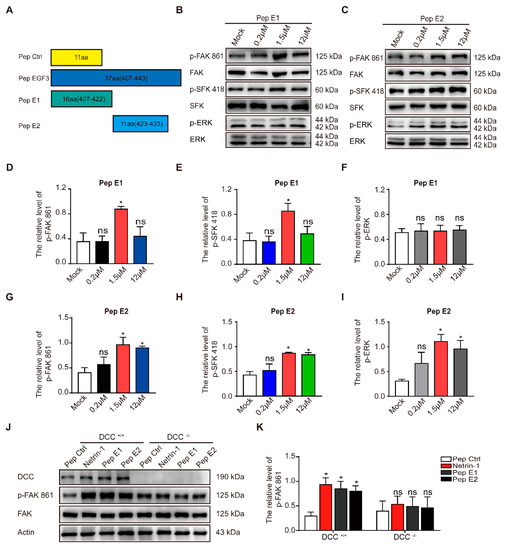
Figure 3.
A
B
C
D
F
B
n
G
I
C
n
J
K
J
n
p
To determine whether the activation of the FAK signaling pathway is DCC-dependent, we measured the extent of tyrosine phosphorylation of FAK induced by these peptides in DCC
+/+
and DCC
−/−
neurons. We found that both peptides E1 and E2 failed to induce the phosphorylation of FAK in DCC
−/−
neurons (
J,K). We also found that synthesized N-terminally Flag-tagged peptide E1 and E2 (Flag-E1 and Flag-E2) direct bind with DCC-FN5 domain (
).
5. Discussion
Our study identified a core peptide sequence of Netrin-1 that is crucial to functional recovery after ICH. By interacting with DCC, this Netrin-1-derived peptide protected against neuronal death due to secondary damage after ICH and is thus a promising treatment for ICH.
The therapeutic application of Netrin-1, a high-molecular-weight protein, requires identification of the minimal functional sequence of Netrin-1 that promotes downstream signaling. In our study, peptides E1 (residues 407–422) and E2 (residues 423–433), which are derived from the EGF3 domain of Netrin-1, respectively, were generated. We found that peptide E1 (residues 407–422) protected against neuronal death after ICH.
We found that Netrin-1 and DCC expression levels were increased in NLT cells after hemin treatment. Hemin is released from hemoglobin after ICH and induces secondary injury to both glia and neuronal cells [41][42][43]. Additionally, we also observed increased protein levels of Netrin-1 and DCC following experimental ICH. Additionally, we also observed increased protein levels of netrin-1 and DCC following experimental ICH. These results are consistent with previous results showing that netrin-1 plays an important role in recovery after ICH [44].
We found that Netrin-1 and DCC expression levels were increased in NLT cells after hemin treatment. Hemin is released from hemoglobin after ICH and induces secondary injury to both glia and neuronal cells [46,47,48]. Additionally, we also observed increased protein levels of Netrin-1 and DCC following experimental ICH. Additionally, we also observed increased protein levels of netrin-1 and DCC following experimental ICH. These results are consistent with previous results showing that netrin-1 plays an important role in recovery after ICH [49].
Furthermore, we found that peptide E1 specifically activated the FAK and SFK signaling pathways. Therefore, peptide E1 may have highly specific effects and be effective in the recovery process after ICH. Unlike peptide E1, peptide E2 can activate ERK signaling, which promotes neuronal death. This may be caused by a Cx(1–2)Cx(3–4)Tx(0 –1)G motif in E2 which is able to activate ERK signaling pathway [28]. This indicates that different Netrin-1-derived peptides perform different functions.
The main receptors of Netrin-1 are DCC and members of the UNC5 family [45]. The role of DCC and UNC5H2 in ICH remains controversial [46][47]. The sequence of Netrin-1 we identified is critical to the interaction of Netrin-1 with DCC [19]. These results suggest that DCC may participate in Netrin-1-induced functional recovery after ICH. Further studies are needed to investigate the underlying mechanisms of these processes and the related signaling pathways. In future experiments, we will assess the biological properties of peptide E1 to explore its toxicity and half-life for clinical application.
The main receptors of Netrin-1 are DCC and members of the UNC5 family [50]. The role of DCC and UNC5H2 in ICH remains controversial [51,52]. The sequence of Netrin-1 we identified is critical to the interaction of Netrin-1 with DCC [19]. These results suggest that DCC may participate in Netrin-1-induced functional recovery after ICH. Further studies are needed to investigate the underlying mechanisms of these processes and the related signaling pathways. In future experiments, we will assess the biological properties of peptide E1 to explore its toxicity and half-life for clinical application.