The receptor tyrosine kinases (RTKs) are a large family of cell-surface receptors, which are essential components of signal transduction pathways. There are more than fifty human RTKs that can be grouped into multiple RTK subfamilies. RTKs mediate cellular signaling transduction, and they play important roles in the regulation of numerous cellular processes. The dysregulation of RTK signaling is related to various human diseases, including cancers. The proteolytic cleavage phenomenon has frequently been found among multiple receptor tyrosine kinases. More and more information about proteolytic cleavage in RTKs has been discovered, providing rich insight.
- receptor tyrosine kinase
- proteolytic cleavage
- protease
- metalloprotease
- secretase
- caspase
- cancers
1. Introduction
Cell-surface receptors play a critical role in cell–cell interactions and the interactions between cells and their microenvironment. Receptor protein kinases are a large family of cell-surface receptors, which include more than 50 members (Figure 1) [1][2][3]. As cell-surface receptors, receptor tyrosine kinases (RTKs) play critical roles in signaling transmission across the plasma membrane. Therefore, RTKs play crucial roles in the regulation of many cellular processes [1][2][3]. Dysregulation of RTKs (such as aberrant activation of RTKs), which causes aberrant RTK signaling, could lead to the development and progression of diseases, such as cancers and diabetes [1][2][3][4][5][6]. Previous research has already found that aberrant activation of RTKs and their downstream signaling pathways play important roles in several cancers. RTKs’ aberrant activation could be caused by several different mechanisms, which include aberrant forms of RTKs (gain of function mutation, truncated mutation etc.), chromosomal rearrangements, genomic amplification, autocrine activation, and aberrant expression of RTK.
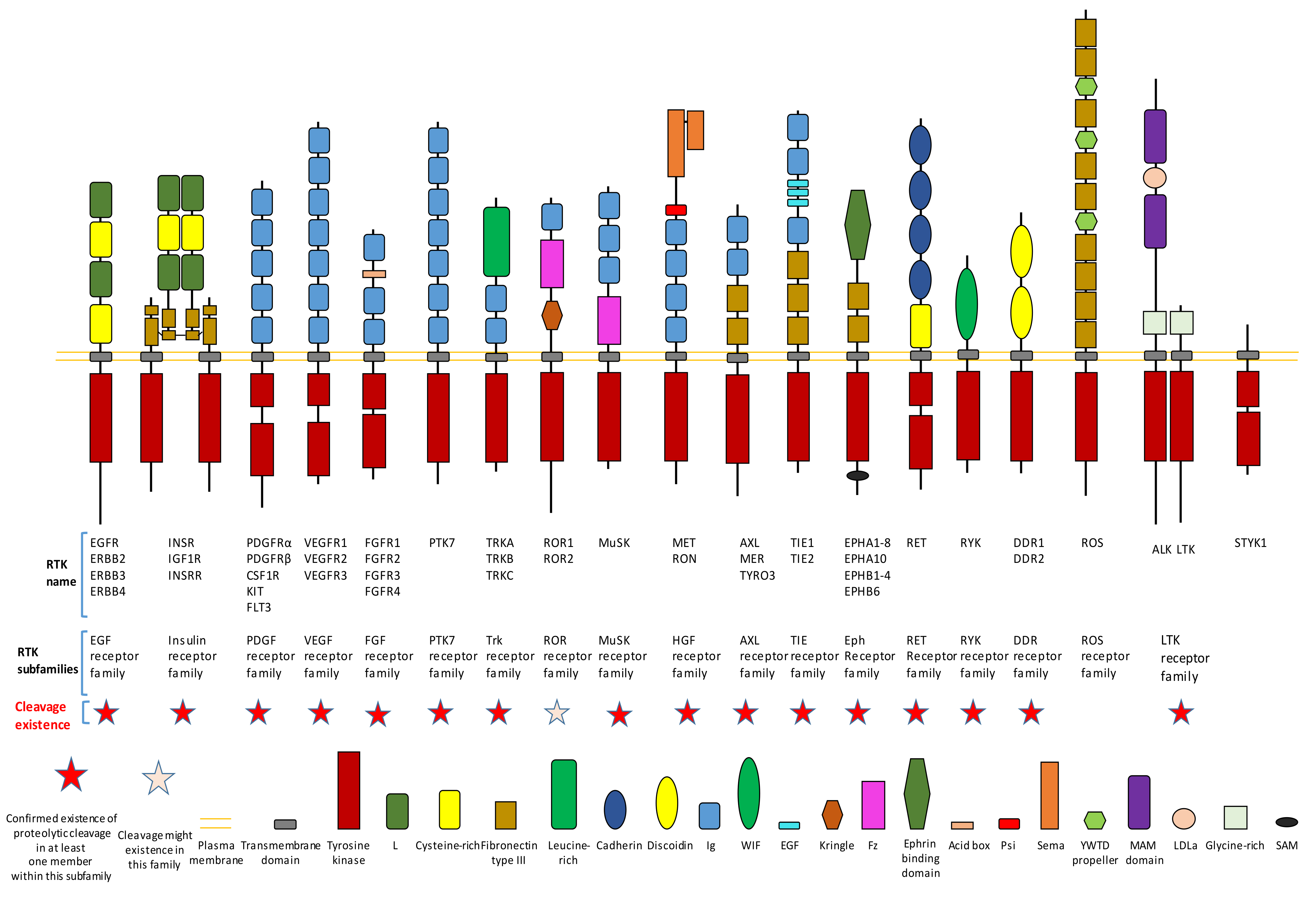 Figure 1. Existence of proteolytic cleavage in different subfamily of receptor tyrosine kinases.
Figure 1. Existence of proteolytic cleavage in different subfamily of receptor tyrosine kinases.
It is crucial to regulate the function of RTKs since they play critical roles in signaling transduction. The regulation of RTKs function can be achieved through various mechanisms. The goal can be achieved in the following ways: mechanisms (such as transcription regulationand translation regulation) that regulate the expression of a receptor; mechanisms that affect the stability, recycling, and degradation of a receptor; the ligand-dependent activation of kinase activity; the ligand-independent activation of kinase activity; and the proteolytic cleavage of receptors. This review will focus on the introduction of proteolytic cleavage in different members of RTK. Hydrolysis of peptide bonds by proteases causes irreversible proteolytic cleavage of proteins and peptides. Proteolytic cleavage of proteins can not only affect their structure, biological function, stability, but also may influence their subcellular location and protein–protein interaction. Specific proteolytic cleavage at a particular site of protein could be viewed as a precision regulation mechanism during posttranslational modification. We must emphasize that, besides the cleavage caused by protease, other mechanisms that can also generate an intracellular truncated RTK include alternative splicing and alternative translation initiation. The objective of this review is to summarize knowledge related to RTK cleavage. This review summarizes information about the relation between RTK cleavage and cancer since cleavage may provide a alternative opportunity for targeting specific RTK in the future. This review also presents information about the cleavage of individual RTK members to better elucidate this phenomenon. After proteolytic cleavage, there are several possible consequences for those truncated fragments, as shown in Figure 2.
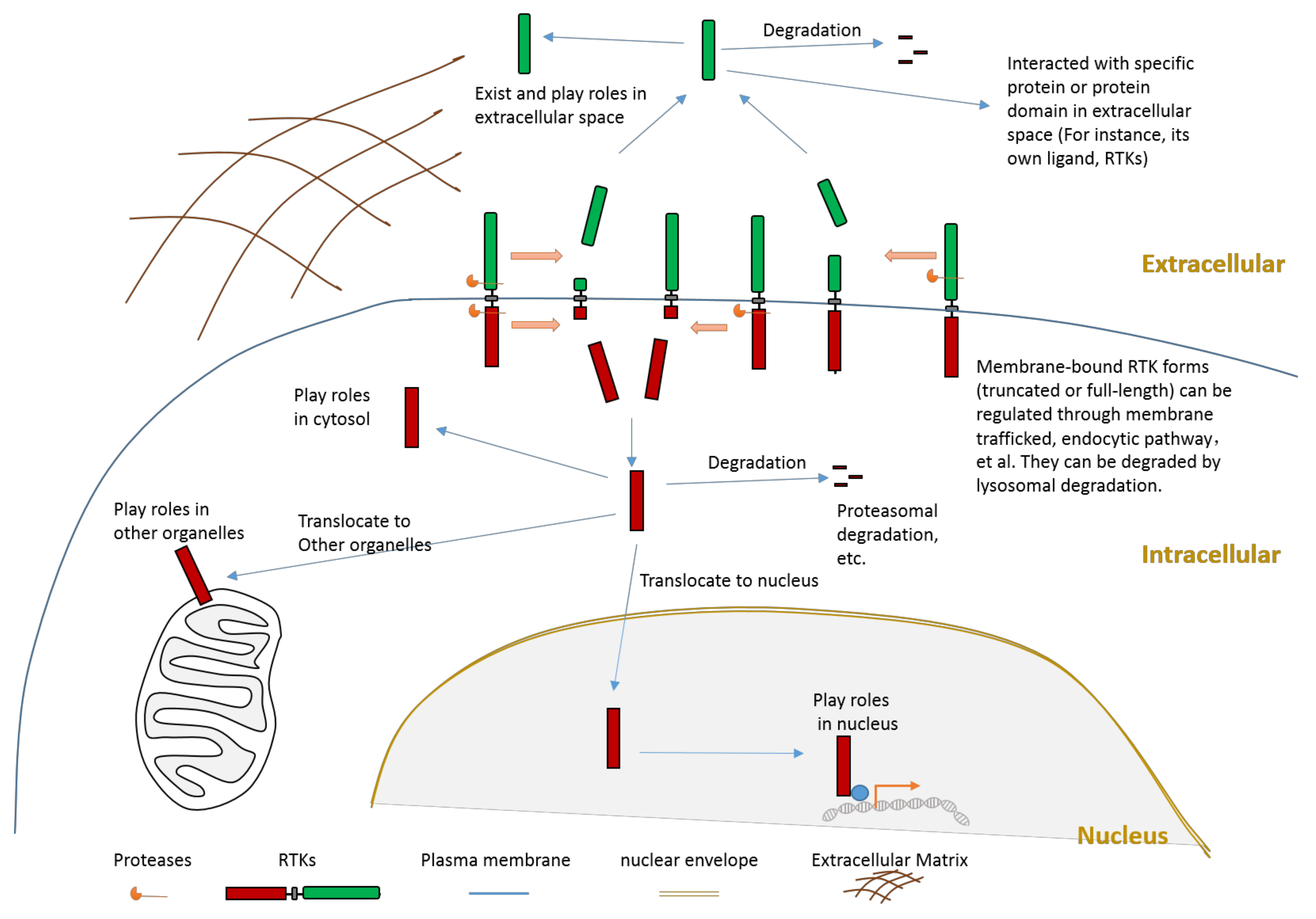
Figure 2. Schematic illustration about the consequences of RTK proteolytic cleavage.
2. Proteolytic Cleavage in Various Members of RTK
Proteolytic cleavage of RTKs can usually release some parts of its intracellular domain (RTK-ICD) or some parts of its extracellular domain (RTK-ECD) (Figure 2). After releasing from the membrane, the RTK-ECDs are shed into extracellular spaces (Figure 2). After being released from the membrane, the RTK-ICDs enter the cytoplasm. Released RTK-ICDs may be translocated into specific subcellular locations, such as the nucleus (Figure 2). Those RTK-ICDs can display their biological function as signaling molecules through distinct mechanisms. These mechanisms include, but are not limited to, interacting with transcriptional factors as co-activators, and phosphorylating target proteins. Some RTK-ICDs, such as EPHB2-ICD, can act as active signaling molecules in the cytoplasm [7]. Many RTK-ICDs, such as RYK-ICD and AXL-ICD, can translocate into the nucleus after cleavage. Moreover, for a few RTKs, the intact receptor can also be translocated to the nucleus. RTK-ICDs can also be degraded after release.
Proteolytic cleavage commonly exists in different subfamilies of RTK (Figure 1). Until now, previous research has already shown that most subfamilies of RTK contain at least one member which has the proteolytic cleavage phenomenon (Figure 1).
2.1. AXL
AXL can be cleaved by proteases ADAM10, ADAM17, and γ-secretase [8][9][10][11] (Figure 3). Cleavage of AXL by ADAM10 and ADAM17 can release its ectodomain from the plasma membrane [8][9][10][11] (Figure 3). Cleavage of AXL by γ-secretase can release an AXL fragment from membrane-anchored AXL into the cytoplasm [10][11] (Figure 3). Previous studies suggested that the cleavage site of γ-secretase is located at the region between Tyr452 and Val459 [10][11]. The ADAM10-mediated mouse Axl cleavage site has been identified within the region between Gln426 and Ala439 (426QPLHHLVSEPPPRA439) [9]. As AXL-ICD contains a nuclear localization sequence (NLS), it could be translocated into the nucleus [10][11]. Therefore, after intracellular cleavage of AXL, AXL-ICD exists both in the cytoplasm and the nucleus.
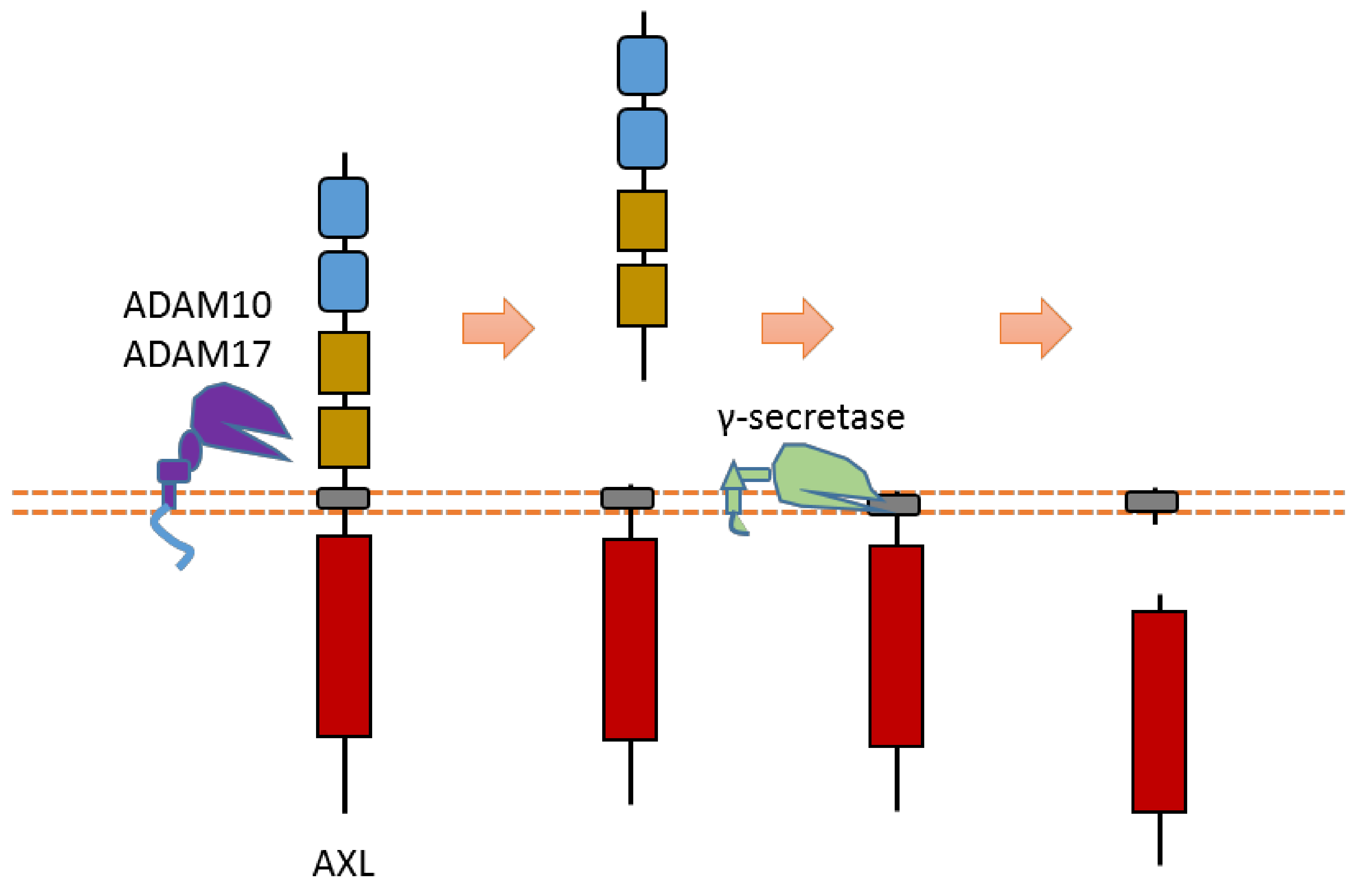
Figure 3. Schematic illustration of proteolytic cleavage in AXL.
2.2. MET
MET is a heterodimer composed of a 50 kDa highly glycosylated α-chain subunit and 145 kDa β-chain (Figure 4). It could be cleaved in multiple positions (Figure 4). Abnormal MET activation plays an important role in a variety of cancers.
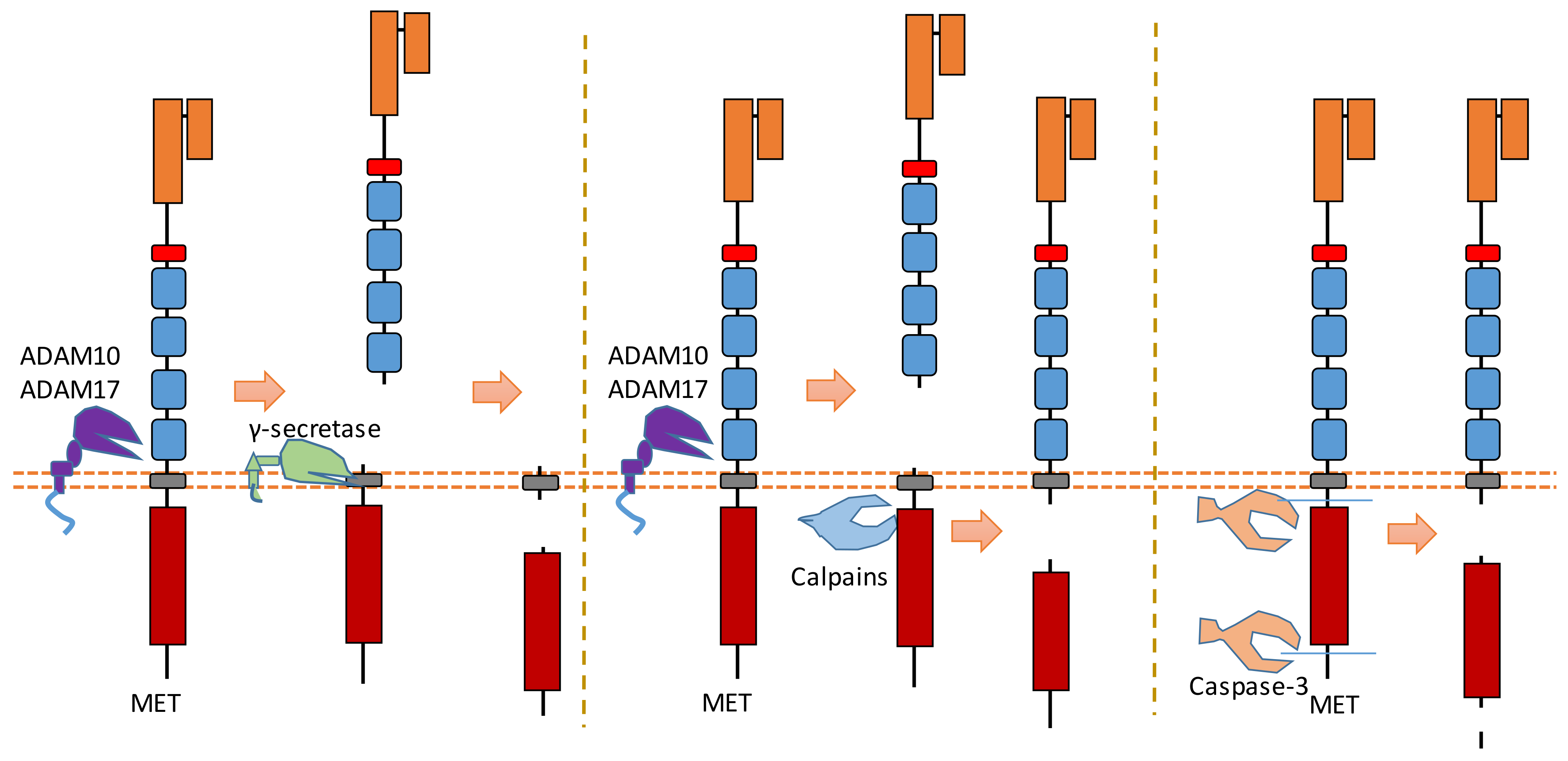
Figure 4. Schematic illustration of proteolytic cleavage in MET.
2.3. Trk A, B, C
All three members (Trk A, B, C) of the Trk subfamily have proteolytic cleavage phenomenon.As one member of the dependence receptor, TrkC is a substrate of caspase-3 (Figure 5). The caspase-dependent cleavage sites are located at Asp495-Asn496 and Asp641-Asn642 [12] (Figure 5). Caspase-dependent TrkC cleavage can release a pro-apoptotic domain, which can enhance apoptosis [12]. In the absence of ligand binding, TrkC can trigger apoptosis. TrkC’s pro-apoptotic activity caused by caspase-dependent TrkC cleavage may mediate sensory neuron death [12].
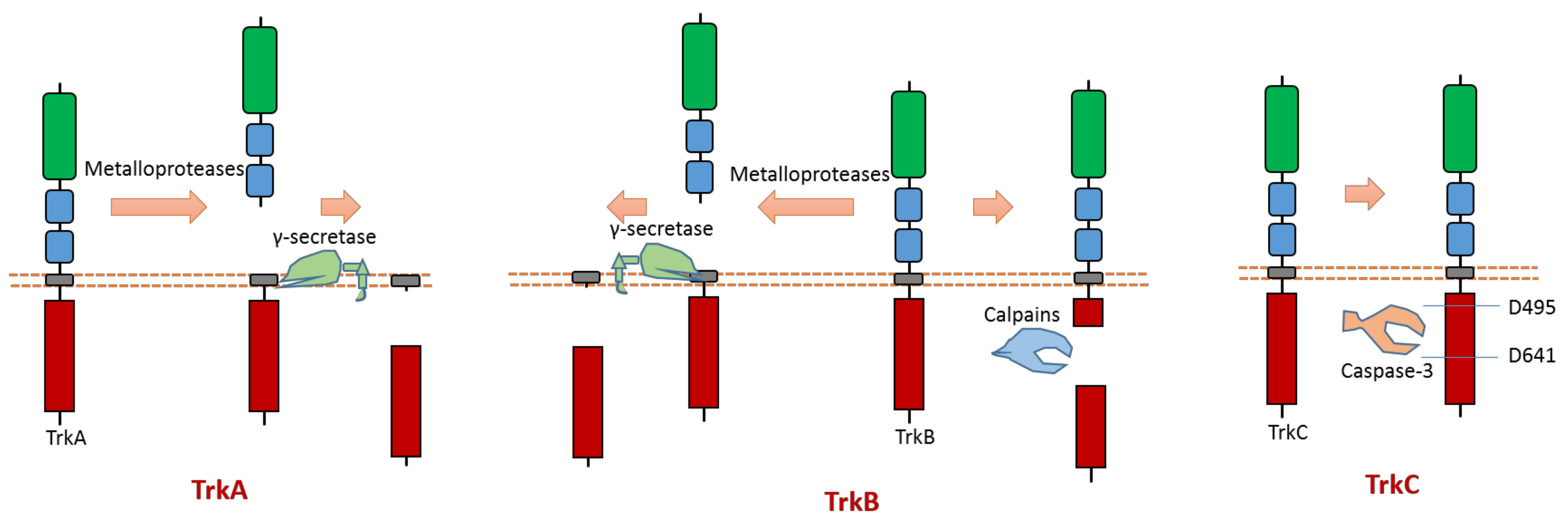
Figure 5. Schematic illustration of proteolytic cleavage in members of Trk subfamily.
2.4. EphA2
As the largest human RTK subfamily, the Eph tyrosine kinase receptors subfamily contains more than ten members. Usually, they can regulate cell–cell and cell–ECM interaction [13]. Among them, the extracellular cleavage of EphA2 has been well studied. EphA2 is found to be expressed in several cancers [14][15][16][17]. EphA2 signaling can affect the cell–cell interaction and motility of cancer cells.
Multiple proteases can cause EphA2 ectodomain cleavage (Figure 6). One study found that EphA2 can be cleaved by membrane type-1 matrix metalloproteinase (MT1-MMP) (Figure 6). The cleavage sites are at Ser431-Val432 and Ile434-Asn435, which are located at the Fibronectin type-III domain [18]. Extracellular cleavage of EphA2 by MT1-MMP can enhance cell repulsion and single-cell invasion of cancer cells [18]. Contact repulsion by membrane-associated ephrins and Eph receptors was suggested to be facilitated by ectodomain cleavage.
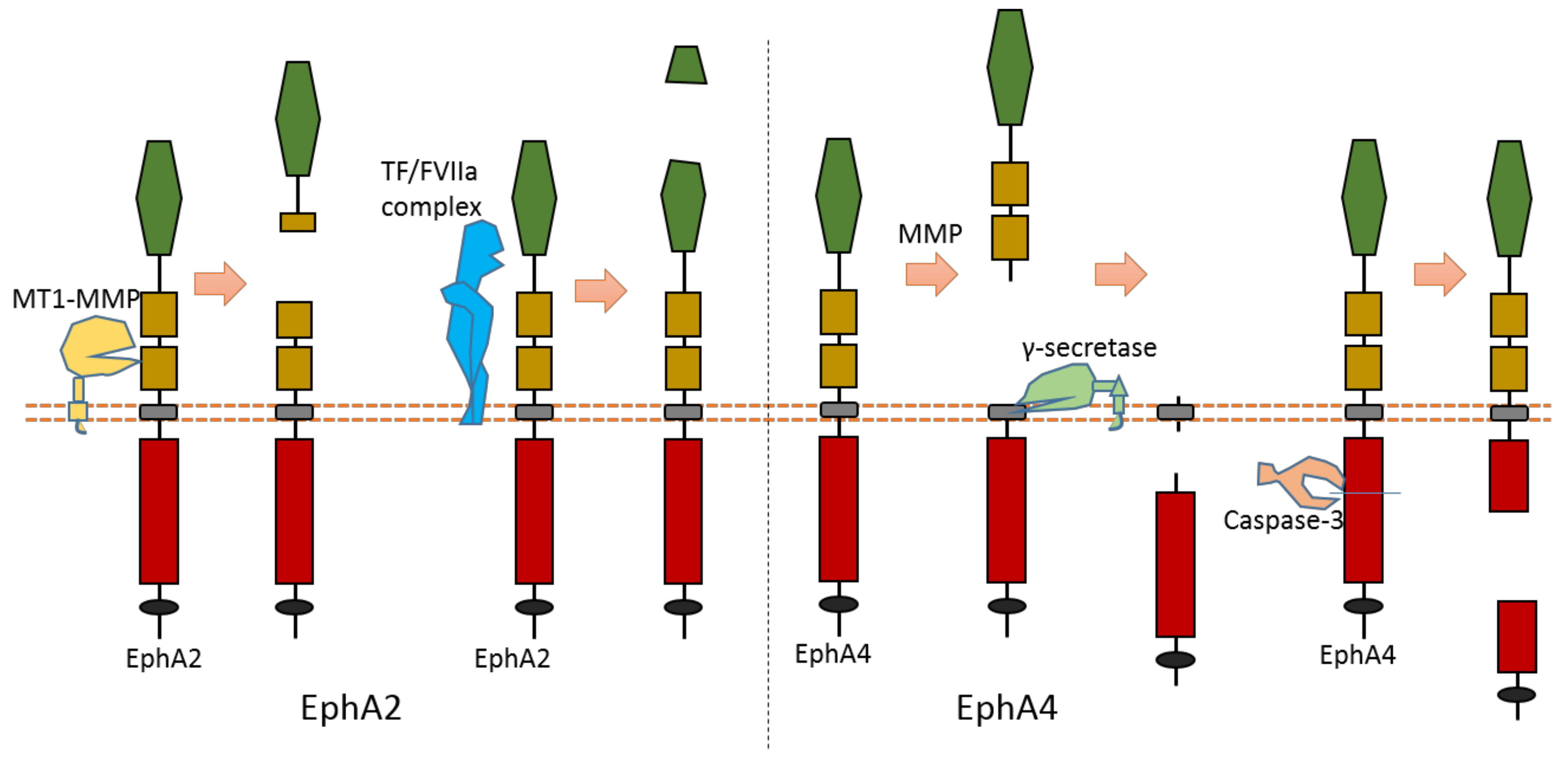
Figure 6. Schematic illustration of proteolytic cleavage in in EphA2 and EphA4.
2.5. EphA4
The cleavage of EphA4 has also been well studied. EphA4 could be cleaved by MMP to release an extracellular fragment [19][20]. EphA4 can be cleaved by γ-secretase to release an intracellular fragment [19][20] (Figure 6). Deletion of one juxtamembrane region (Asn533-Thr547) with 15 amino acids in EphA4 can cause resistance to cleavage, which suggests that the cleavage site is located at this region [19][20]. One research study found that ECD cleavage of EphA4 is temporally and spatially regulated during development [20]. ECD cleavage of EphA4 was found to affect cell–cell interaction and motor axon guidance [20]. EphA4 cleavage can regulate the balance of receptor–ligand interactions, which are the interactions between EphA4 and ephrin-A5. EphA4 cleavage can mediate the availability of free ephrin-As in the axon intermediate target, which is crucial to axon guidance in vivo [20]. Previous research showed that mice with blocked EphA4 cleavage have motor axon guidance defects in a mouse model [20]. Research also found that blocking EphA4 cleavage increased the expression of full-length EphA4 in limb mesenchyme. Additionally, a previous study also suggested that EphA4 can associate with ADAM10 to form a complex with ADAM10 and E-cadherin in the plasma membranes of pillar cells [21]. This study suggests that EphA4 promotes the destruction of E-cadherin-based adhesions between adjacent pillar cells [21]; and that, with the presence of Ephrin-B2, EphA4 can promote the activation of ADAM10, which results in the cleavage of E-cadherin [21]. In addition, EphA4 is a member of dependence receptor [22]. EphA4 can be cleaved by caspase-3 at Asp774-Pro775 to enhance apoptosis in the absence of ligand [22].
2.6. DDR1
DDR family RTKs (DDR1 and DDR2) play critical roles in cell–ECM interaction [23][24]. Some collagens can bind to DDRs as ligands to induce DDR receptor phosphorylation, which then can activate the DDR signaling transduction [23][24]. Therefore, DDRs, which are involved in the communication of cells and the extracellular microenvironment, can regulate cell–ECM adhesion, cell migration, and some other biological processes.
Some membrane-bound protease MT-MMPs (MT1-MMP, MT2-MMP, and MT3-MMP) could cleave DDR1 in its ECD region, which can attenuate collagen I- and IV-induced receptor phosphorylation [25][26] (Figure 7). MT1-MMP can cleave DDR1 at its extracellular juxtamembrane region, which generates a membrane-bound C-terminal fragment and an N-terminal fragment [25][26]. The cleavage sites were identified at Ser397-Leu398 and Pro407-Val408 [25][26]. In addition, some research found that ADAM10 also can interact with DDR1 to cleave it when collagen is bound to DDR1 [25][27] (Figure 7).
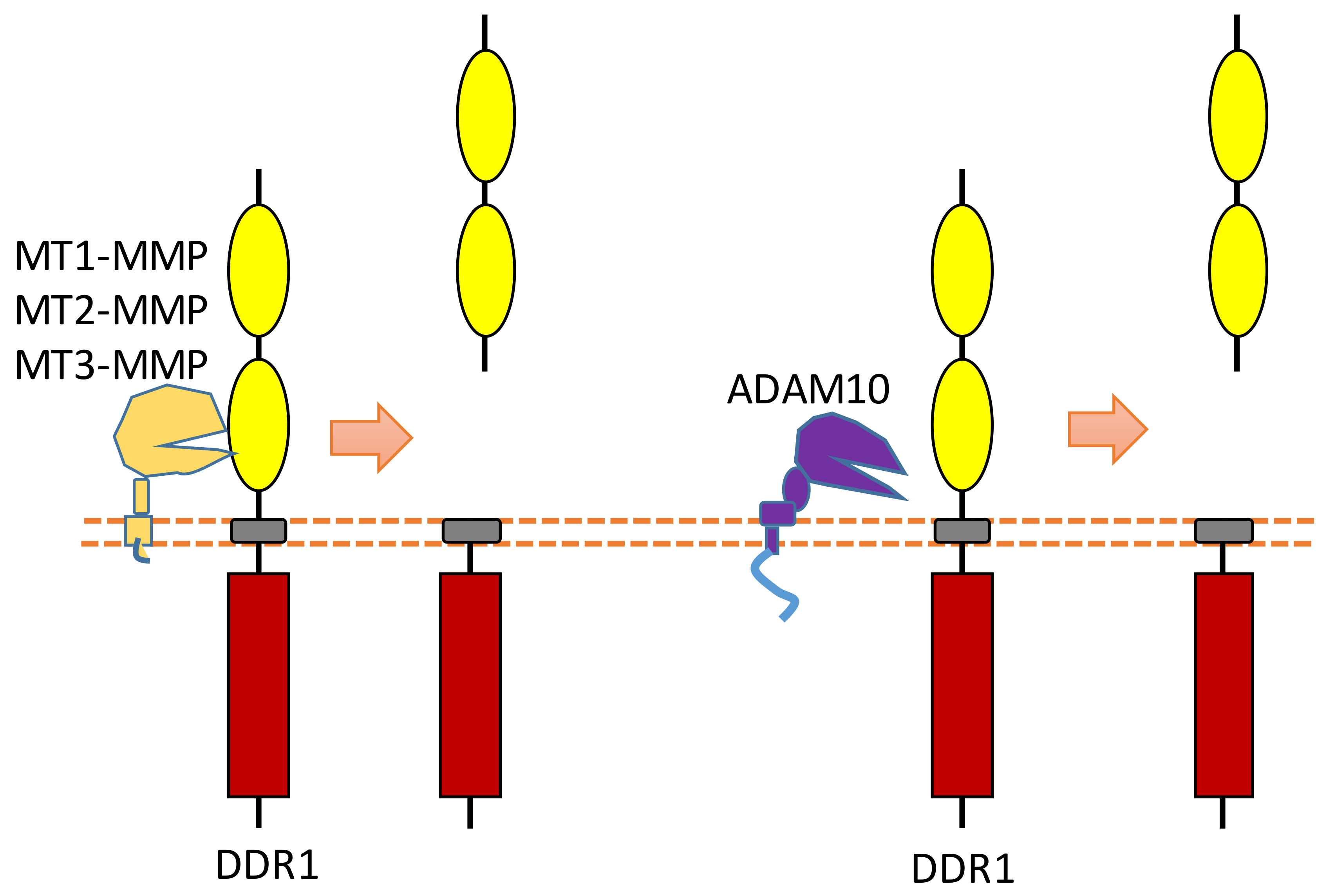
Figure 7. Schematic illustration of proteolytic cleavage in DDR.
3. Summary
Proteolytic cleavage is a common phenomenon that had already been found in many different RTK members. RTK cleavage research is an emerging research field. Systematic investigation of this biological process will help us to understand RTKs better. Most RTK cleavage is conducted by specific proteases at specific cleavage positions; therefore, regulation of RTK cleavage may be achieved through several mechanisms, including protease activity inhibition and regulation of protease activation. Knowledge about RTK cleavage may provide valuable opportunities for targeting RTK cleavage in the future.
References
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134.
- Hubbard, S.R.; Miller, W.T. Receptor tyrosine kinases: Mechanisms of activation and signaling. Curr. Opin. Cell Biol. 2007, 19, 117–123.
- Casaletto, J.B.; McClatchey, A.I. Spatial regulation of receptor tyrosine kinases in development and cancer. Nat. Rev. Cancer 2012, 12, 387–400.
- Taylor, S.I.; Accili, D.; Cama, A.; Kadowaki, H.; Kadowaki, T.; Imano, E.; Sierra, M.L. Mutations in the insulin receptor gene in patients with genetic syndromes of insulin resistance. Adv. Exp. Med. Biol. 1991, 293, 197–213.
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191.
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58.
- Xu, J.; Litterst, C.; Georgakopoulos, A.; Zaganas, I.; Robakis, N.K. Peptide ephb2/ctf2 generated by the gamma-secretase processing of ephb2 receptor promotes tyrosine phosphorylation and cell surface localization of n-methyl-d-aspartate receptors. J. Biol. Chem. 2009, 284, 27220–27228.
- O’Bryan, J.P.; Fridell, Y.W.; Koski, R.; Varnum, B.; Liu, E.T. The transforming receptor tyrosine kinase, axl, is post-translationally regulated by proteolytic cleavage. J. Biol. Chem. 1995, 270, 551–557.
- Orme, J.J.; Du, Y.; Vanarsa, K.; Mayeux, J.; Li, L.; Mutwally, A.; Arriens, C.; Min, S.; Hutcheson, J.; Davis, L.S.; et al. Heightened cleavage of axl receptor tyrosine kinase by adam metalloproteases may contribute to disease pathogenesis in sle. Clin. Immunol. 2016, 169, 58–68.
- Lu, Y.; Wan, J.; Yang, Z.; Lei, X.; Niu, Q.; Jiang, L.; Passtoors, W.M.; Zang, A.; Fraering, P.C.; Wu, F. Regulated intramembrane proteolysis of the axl receptor kinase generates an intracellular domain that localizes in the nucleus of cancer cells. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 1382–1397.
- Merilahti, J.A.M.; Ojala, V.K.; Knittle, A.M.; Pulliainen, A.T.; Elenius, K. Genome-wide screen of gamma-secretase-mediated intramembrane cleavage of receptor tyrosine kinases. Mol. Biol. Cell 2017, 28, 3123–3131.
- Tauszig-Delamasure, S.; Yu, L.Y.; Cabrera, J.R.; Bouzas-Rodriguez, J.; Mermet-Bouvier, C.; Guix, C.; Bordeaux, M.C.; Arumae, U.; Mehlen, P. The trkc receptor induces apoptosis when the dependence receptor notion meets the neurotrophin paradigm. Proc. Natl. Acad. Sci. USA 2007, 104, 13361–13366.
- Nievergall, E.; Lackmann, M.; Janes, P.W. Eph-dependent cell-cell adhesion and segregation in development and cancer. Cell. Mol. Life Sci. Cmls 2012, 69, 1813–1842.
- Tandon, M.; Vemula, S.V.; Mittal, S.K. Emerging strategies for epha2 receptor targeting for cancer therapeutics. Expert Opin. Ther. Targets 2011, 15, 31–51.
- Walker-Daniels, J.; Coffman, K.; Azimi, M.; Rhim, J.S.; Bostwick, D.G.; Snyder, P.; Kerns, B.J.; Waters, D.J.; Kinch, M.S. Overexpression of the epha2 tyrosine kinase in prostate cancer. Prostate 1999, 41, 275–280.
- Zelinski, D.P.; Zantek, N.D.; Stewart, J.C.; Irizarry, A.R.; Kinch, M.S. Epha2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res. 2001, 61, 2301–2306.
- Martini, G.; Cardone, C.; Vitiello, P.P.; Belli, V.; Napolitano, S.; Troiani, T.; Ciardiello, D.; Della Corte, C.M.; Morgillo, F.; Matrone, N.; et al. Epha2 is a predictive biomarker of resistance and a potential therapeutic target for improving antiepidermal growth factor receptor therapy in colorectal cancer. Mol. Cancer Ther. 2019, 18, 845–855.
- Kikuchi, K.; Kozuka-Hata, H.; Oyama, M.; Seiki, M.; Koshikawa, N. Identification of proteolytic cleavage sites of epha2 by membrane type 1 matrix metalloproteinase on the surface of cancer cells. Methods Mol. Biol. 2018, 1731, 29–37.
- Inoue, E.; Deguchi-Tawarada, M.; Togawa, A.; Matsui, C.; Arita, K.; Katahira-Tayama, S.; Sato, T.; Yamauchi, E.; Oda, Y.; Takai, Y. Synaptic activity prompts gamma-secretase-mediated cleavage of epha4 and dendritic spine formation. J. Cell Biol. 2009, 185, 551–564.
- Gatto, G.; Morales, D.; Kania, A.; Klein, R. Epha4 receptor shedding regulates spinal motor axon guidance. Curr. Biol. CB 2014, 24, 2355–2365.
- Defourny, J.; Peuckert, C.; Kullander, K.; Malgrange, B. Epha4-adam10 interplay patterns the cochlear sensory epithelium through local disruption of adherens junctions. iScience 2019, 11, 246–257.
- Furne, C.; Ricard, J.; Cabrera, J.R.; Pays, L.; Bethea, J.R.; Mehlen, P.; Liebl, D.J. Ephrinb3 is an anti-apoptotic ligand that inhibits the dependence receptor functions of epha4 receptors during adult neurogenesis. Biochim. Biophys. Acta 2009, 1793, 231–238.
- Vogel, W.; Gish, G.D.; Alves, F.; Pawson, T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol. Cell 1997, 1, 13–23.
- Juskaite, V.; Corcoran, D.S.; Leitinger, B. Collagen induces activation of ddr1 through lateral dimer association and phosphorylation between dimers. eLife 2017, 6, e25716.
- Vogel, W.F. Ligand-induced shedding of discoidin domain receptor 1. FEBS Lett. 2002, 514, 175–180.
- Fu, H.L.; Sohail, A.; Valiathan, R.R.; Wasinski, B.D.; Kumarasiri, M.; Mahasenan, K.V.; Bernardo, M.M.; Tokmina-Roszyk, D.; Fields, G.B.; Mobashery, S.; et al. Shedding of discoidin domain receptor 1 by membrane-type matrix metalloproteinases. J. Biol. Chem. 2013, 288, 12114–12129.
- Shitomi, Y.; Thogersen, I.B.; Ito, N.; Leitinger, B.; Enghild, J.J.; Itoh, Y. Adam10 controls collagen signaling and cell migration on collagen by shedding the ectodomain of discoidin domain receptor 1 (ddr1). Mol. Biol. Cell 2015, 26, 659–673.