Muse cells, identified as pluripotent surface marker, stage-specific embryonic antigen (SSEA)-3(+), are endogenous reparative pluripotent stem cells distributed in the bone marrow, peripheral blood and connective tissue of every organ. Since they are non-tumorigenic and do not require gene introduction or cytokine treatment to be rendered pluripotent and induce differentiation, they elicit few safety concerns. They can be delivered intravenously and do not require surgery for their administration since they selectively home to damaged site by sphingosine-1-phosphate (S1P)-S1PR2 axis after intravenous injection. Donor-Muse cells can be used without HLA-matching test or immunosuppressant treatment since they have a specific immunomodulatory system represented by HLA-G expression.
Muse cells, identified as pluripotent surface marker, stage-specific embryonic antigen (SSEA)-3(+), are endogenous reparative pluripotent stem cells distributed in the bone marrow, peripheral blood and connective tissue of every organ. Since they are non-tumorigenic and do not require gene introduction or cytokine treatment to be rendered pluripotent and induce differentiation, they elicit few safety concerns. They can be delivered intravenously and do not require surgery for their administration since they selectively home to damaged site by sphingosine-1-phosphate (S1P)-S1PR2 axis after intravenous injection. Donor-Muse cells can be used without HLA-matching test or immunosuppressant treatment since they have a specific immunomodulatory system represented by HLA-G expression.
- SSEA-3
- pluripotent
- MSCs
- sphingosine-1-phosphate
- ALS
- stroke
1. Introduction
The body infrastructure requires continuous maintenance; body tissues and organs persistently undergo minute damage, which is rapidly and efficiently repaired to maintain tissue homeostasis throughout life. We take this natural reparative activity of the body for granted, and the molecular details of the reparative mechanisms are not yet fully understood.
Multilineage-differentiating, stress-enduring (Muse) cells are considered an important part of the body maintenance system. Muse cells, identified as pluripotent surface marker, stage-specific embryonic antigen (SSEA)-3(+), are endogenous, reparative, non-tumorigenic, pluripotent stem cells distributed throughout the body [1][2]. They are constantly mobilized from the bone marrow to the peripheral blood and thus supplied to every organ (
Multilineage-differentiating, stress-enduring (Muse) cells are considered an important part of the body maintenance system. Muse cells, identified as pluripotent surface marker, stage-specific embryonic antigen (SSEA)-3(+), are endogenous, reparative, non-tumorigenic, pluripotent stem cells distributed throughout the body [1,2]. They are constantly mobilized from the bone marrow to the peripheral blood and thus supplied to every organ (
Figure 1) [3][4]. Muse cells exhibit pluripotency and are able to differentiate into ectodermal, mesodermal, and endodermal cells and self-renew from a single cell [1]. These beneficial characteristics of Muse cells allow them to differentiate into cells comprising various types of tissue to replenish damaged/lost cells. Tissues comprise a three-dimensional organization of multiple cell types. For tissue repair, Muse cells spontaneously differentiate into multiple cell types comprising the damaged tissue and integrate into the proper position to maintain tissue function [2]. In this manner, they participate in the daily minute repair (
) [3,4]. Muse cells exhibit pluripotency and are able to differentiate into ectodermal, mesodermal, and endodermal cells and self-renew from a single cell [1]. These beneficial characteristics of Muse cells allow them to differentiate into cells comprising various types of tissue to replenish damaged/lost cells. Tissues comprise a three-dimensional organization of multiple cell types. For tissue repair, Muse cells spontaneously differentiate into multiple cell types comprising the damaged tissue and integrate into the proper position to maintain tissue function [2]. In this manner, they participate in the daily minute repair (
). Damaged cells actively produce sphingosine-1-phosphate (S1P) by phosphorylating sphingosine, a cell membrane component, and thus, S1P is the general alert signal of tissue damage [5]. Muse cells express S1P receptor 2 (S1PR2), allowing them to sharply sense the S1P alert signal produced by the damaged tissue and selectively home to the site of damage where they accumulate (
) [6].
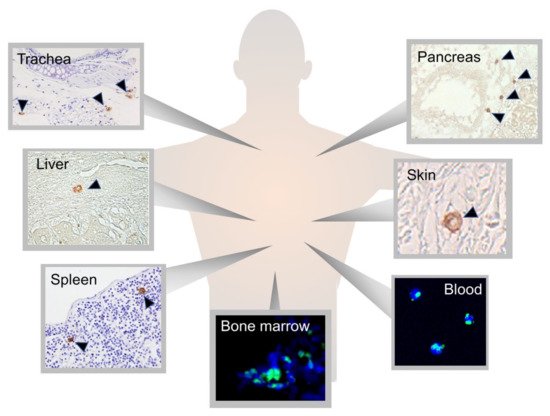
Figure 1.
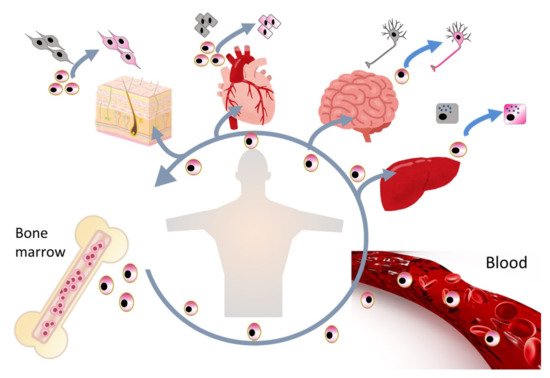
Figure 2.
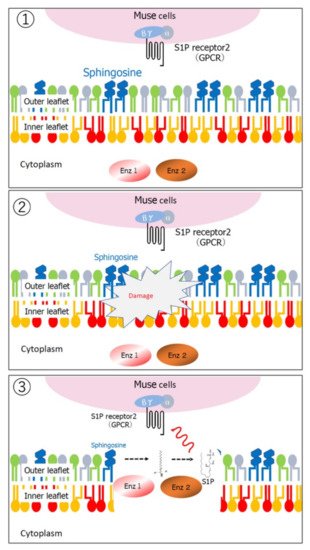
Figure 3.
1
2
3
Endogenous Muse cells thus act as reparative stem cells through the above-described series of reactions. Suppose that extensive tissue damage is caused, such as by a stroke; the post-infarct tissue produces S1P as a damage alert signal, thereby mobilizing endogenous Muse cells from the bone marrow to the circulating blood to travel to the infarct area and repair the affected tissue. Clinical data support this hypothesis; the number of peripheral blood-Muse cells sharply increases after the onset of stroke and acute myocardial infarction [3][7]. In an acute myocardial infarction study, endogenous Muse cell dynamics in the acute phase was shown to play an important role in the prognosis of patients; patients with a higher number of Muse cells in the peripheral blood in the acute phase exhibited statistically meaningful cardiac function recovery with less occurrence of heart failure at 6 months, compared with another group who did not exhibit an increased number of circulating Muse cell during the acute phase, suggesting their innate reparative function for the heart [3][9]. Thus, the number of endogenous Muse cells is a potential parameter of the body’s reparative activity. If the number of Muse cells is insufficient for repair, or if the patient’s endogenous Muse cells have low reparative activity due to underlying diseases, exogenous Muse cells, collectable from the bone marrow, peripheral blood, organ connective tissues, can be supplied via intravenous infusion to strengthen the body’s reparative activity (
Endogenous Muse cells thus act as reparative stem cells through the above-described series of reactions. Suppose that extensive tissue damage is caused, such as by a stroke; the post-infarct tissue produces S1P as a damage alert signal, thereby mobilizing endogenous Muse cells from the bone marrow to the circulating blood to travel to the infarct area and repair the affected tissue. Clinical data support this hypothesis; the number of peripheral blood-Muse cells sharply increases after the onset of stroke and acute myocardial infarction [3,7]. In an acute myocardial infarction study, endogenous Muse cell dynamics in the acute phase was shown to play an important role in the prognosis of patients; patients with a higher number of Muse cells in the peripheral blood in the acute phase exhibited statistically meaningful cardiac function recovery with less occurrence of heart failure at 6 months, compared with another group who did not exhibit an increased number of circulating Muse cell during the acute phase, suggesting their innate reparative function for the heart [3,9]. Thus, the number of endogenous Muse cells is a potential parameter of the body’s reparative activity. If the number of Muse cells is insufficient for repair, or if the patient’s endogenous Muse cells have low reparative activity due to underlying diseases, exogenous Muse cells, collectable from the bone marrow, peripheral blood, organ connective tissues, can be supplied via intravenous infusion to strengthen the body’s reparative activity (
). This is the basic concept of Muse cell therapy.
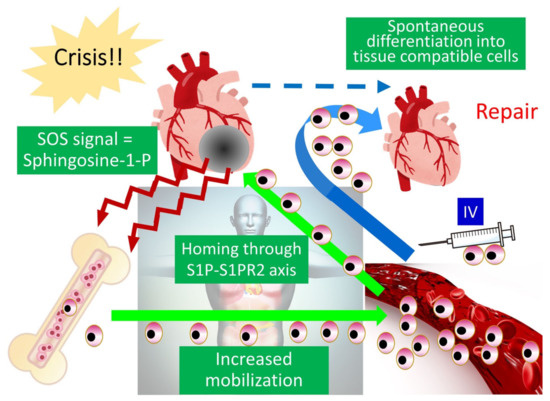
Figure 4.
Another unique characteristic of Muse cells, compared with other stem cells is their unique immune privilege system. Donor-derived (allogenic) Muse cells do not require human leucocyte antigen (HLA)-matching or long-term immunosuppressant treatment [6]. Thus, on the basis of these characteristics, Muse cells provide simple, sophisticated next-generation medical care that can be implemented not only in advanced medical institutions but also in general hospitals and clinics due to the following advantages:
-
Muse cells are endogenous and therefore elicit few safety concerns.
- Muse cells can be delivered intravenously and do not require surgery for their administration.
- Muse cells do not require gene introduction or cytokine treatment to be rendered pluripotent and induce differentiation.
- Donor Muse cells can be used without HLA-matching or immunosuppressant treatment.
- Muse cells remain incorporated as functional cells in the host tissue for an extended period of time, making their anti-inflammatory, anti-apoptotic, and trophic effects long-lasting.
Clinical trials in which donor Muse cells are delivered by intravenous infusion without HLA-matching and immunosuppressant treatment to patients with acute myocardial infarction [10], stroke, epidermolysis bullosa [11], spinal cord injury, neonatal cerebral palsy, and amyotrophic lateral sclerosis (ALS) are in progress (
).
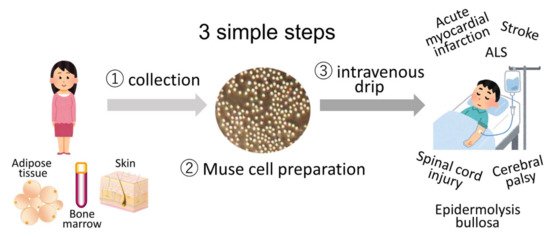
Figure 5.
Strategy for Muse cell clinical trials consists of 3 simple steps. Muse cells, collectable from donor sources such as the bone marrow, adipose tissue, and skin, are expanded to produce Muse cell preparations and directly delivered to patients with acute myocardial infarction, stroke, epidermolysis bullosa, spinal cord injury, cerebral palsy, and ALS by intravenous drip without HLA-matching and immunosuppressant treatment.
2. Basic Characteristics of Muse Cells
2.1. Muse Cells as Endogenous Reparative Stem Cells Are Widely Distributed in the Body
Muse cells are identified as cells positive for stage-specific embryonic antigen (SSEA)-3, a representative marker of pluripotent stem cells [1]. SSEA-3 is an antibody that recognizes a sugar epitope on the cell surface of pluripotent/totipotent cells, such as embryonic stem (ES) cells and epiblast stem cells in the two-cell stage during normal development [12][13]. Because sugar is the epitope recognized by the SSEA-3 antibody, there is no gene that directly encodes SSEA-3; thus, specific knockout animals for SSEA-3 are difficult to generate. On the other hand, species differences in SSEA-3 do not exist, and SSEA-3 can thus be applied to identify Muse cells across species. In fact, Muse cells have been identified in several mammals, including mouse, rat, rabbit, goat, and swine, using SSEA-3 [6][14][15][16][17].
Figure 1) [8][18]. They are also found in extra-embryonic tissues such as the umbilical cord, which is rich in connective tissue [19]. Muse cells comprise approximately ~0.03% (1:3000) of mononucleated cells in the bone marrow and form loose clusters near blood vessels [1][7]. They are constantly mobilized from the bone marrow to the peripheral blood at a rate of approximately 0.01~0.2% of the mononucleated cell fraction in the blood. Large individual differences exist, however, and the rate varies greatly even within individuals, depending on the physical condition and presence of illness or injury [3][4].
Muse cells positive for SSEA-3 can be recognized in connective tissue. Somatic stem cells are known to have their own niche, an area of a tissue that provides a specific microenvironment in which stem cells maintain a quiescent state, e.g., hair follicle and hematopoietic stem-cell niches [20][21]. To date, Muse cells have not been observed in any particular niche-like tissue structures. Rather, they are freely and sparsely distributed in the connective tissue, probably due to their continuous active dynamic movement in vivo [18].
Besides SSEA-3, Muse cells express the pluripotent master genes Oct3/4, Nanog, and Sox2, as well as other pluripotency-related markers, such as Rex1, PAR4, BMP4, CBX7, DAZL, DPPA3, DPPA4, FGFR1, GDF3, KLF4, MSX2, Myc, NR0B1, Prdm1, Six4, SPRY1/2, SSBP2, and UTF1 [18]. A single Muse cell can generate cells representative of all three germ layers; in suspension culture, single Muse cells form embryoid-like clusters [1]. The cluster cells spontaneously generate endodermal (positive for GATA-6, cytokeratin 7, and alpha-fetoprotein), mesodermal (positive for Nkx2.5, smooth muscle actin, and desmin), and ectodermal (positive for MAP-2 and neurofilament) cells without any cytokine induction when transferred onto gelatin-coated culture dishes for expansion [1]. Such triploblastic differentiation from a single cell can be reproduced over generations, indicating self-renewability. In addition to their spontaneous differentiation, Muse cells also differentiate in vitro at a high rate (~80–95%) into various target cell types, such as hepatic-, cardiac- and neural-lineage cells, as well as into adipocytes, osteocytes, keratinocytes, and melanocytes, when certain sets of cytokines are supplied in a step-wise manner [22][23][24]. Because of these two core characteristics, triploblastic differentiation and self-renewability at a single cell level, Muse cells are considered pluripotent stem cells.
2.2. Sources of Muse Cells
Muse cells are collectable as SSEA-3(+) cells from various sources, such as the bone marrow, peripheral blood, and connective tissue of nearly every organ, including extraembryonic tissues, such as the umbilical cord [1][18][19]. Importantly, commercially available cultured mesenchymal stem cells (MSCs) established from bone marrow, adipose tissue, and umbilical cord, as well as from dermal fibroblasts, are also practical sources of Muse cells [25]. Several percent of the total population of MSCs and fibroblasts can be collected as SSEA-3(+) Muse cells [22].
2.3. Stress Tolerance, High DNA Repair Ability, and Non-Tumorigenicity
2
2
Although Muse cells exhibit pluripotency, they have low telomerase activity, comparable to that of somatic cells, and do not form teratomas for up to 6 months when transplanted into the testis of immunodeficient mice [22][26][30]. In fact, gene expression levels of factors relevant to cell cycle progression in Muse cells are similar to those in somatic cells and substantially lower than those in ES and induced pluripotent stem (iPS) cells [22]. On the other hand, the proliferation speed of Muse cells is ~1.3 days/cell division, similar to or slightly slower than that of fibroblasts, and they are thus expandable to clinical scale [1]. Thus, Muse cells are pluripotent, endogenous, and non-tumorigenic.
2.4. Ability to Selectively Home to Sites of Damage
Escherichia coli (STEC)-associated encephalopathy [37], as well as in rat models of middle cerebral artery occlusion ischemia and perinatal hypoxic ischemic encephalopathy [38][39], Muse cells exhibited superiority over MSCs/non-Muse MSCs in selective homing to the sites of damage. Not only in these animal models, but also the data collected from patients with stroke and acute myocardial infarction demonstrated that an increase in the serum S1P level precedes the increase in the number of circulating endogenous Muse cells after the cell injury onset [3][7]. These findings indicate that the S1P–S1PR2 axis is the key system that controls the selective homing of circulating Muse cells to sites of damage.
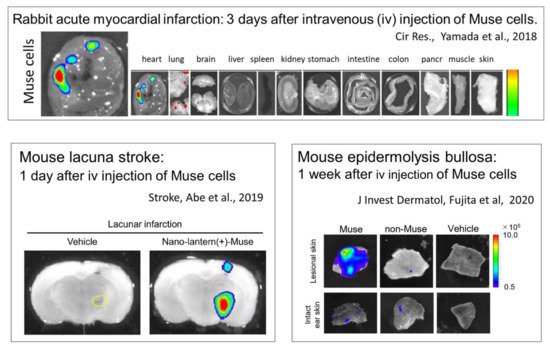
Figure 6.
2.5. Replacement of Damaged/Apoptotic Cells by Spontaneous Differentiation of Muse Cells into the Damaged/Apoptotic Cell Type
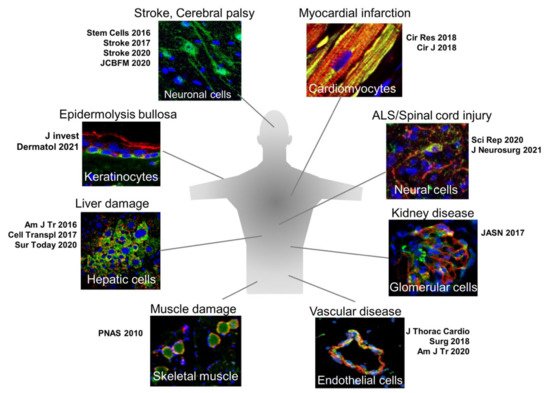
Figure 7.
Another characteristic is that in vivo differentiation proceeds rapidly, compared with in vitro cytokine-induced differentiation. When proper sets of cytokines are supplied in vitro, more than 80% of Muse cells differentiate into melanocytes, cardiomyocytes, osteocytes, adipocytes, and neural- and hepatic-lineage cells, but generally it takes at least several months [22][23][24]. In vivo, however, Muse cells that homed to the post-infarct area of a stroke model spontaneously differentiated into neuronal cells (~60% of engrafted cells) and oligodendrocytes (12~25%) with a rapid time course: elongated neurites and expressed progenitor markers NeuroD and Mash1 within 3 days, forming a network-like structure, and expressed maturity markers MAP2 and NeuN at 7 days [34][38]. Muse cell-derived neuronal cells incorporate into the pyramidal tract, including the pyramidal decussation, as demonstrated by anterograde and retrograde tracing, and into the sensory tract, as demonstrated by somatosensory-evoked potentials and the formation of synapses with host neuronal cells at 3 months, leading to statistically meaningful functional recovery [34][38]. Spontaneous differentiation of Muse cells into neuronal and glial cells after homing to the damaged central nervous system is also reported in other models, perinatal hypoxic ischemic encephalopathy, brain hemorrhage, ALS, and STEC-related encephalopathy [36][37][39][40]. In an acute myocardial infarction model, Muse cells homed to the post-infarct tissue and within 2 weeks spontaneously differentiated into cells positive for cardiomyocyte markers, such as troponin-I, sarcomeric α-actinin, and connexin 43, exhibiting calcium influx and efflux synchronous with heart activity recorded by an electrocardiogram [6].
In mouse liver damage models, human Muse cells expressed CK19, DLK, OV6, and alpha-fetoprotein, markers of liver progenitor cells, at 2 days after intravenous injection and expressed mature hepatocyte markers HepPar1, albumin, and anti-trypsin within 2 weeks [27][41]. Muse cell-derived hepatocytes that did not fuse with host hepatocytes in fluorescence in situ hybridization expressed cytochrome P450, family 1, subfamily A, polypeptide2, and glucose-6-phosphatase, enzymes related to drug metabolism and glycolysis, and delivered increased serum albumin and decreased total bilirubin levels, suggesting that the Muse cells were functioning as hepatocytes [27][41]. The same tendency was reproduced in a swine hepatectomy model intravenously injected with allogenic-Muse cells [16].
2.6. Immune Privilege of Muse Cells
Immunologic rejection is a main drawback of allogeneic-based cell therapy [43]. Based on their immunomodulatory effects, MSCs are applied for graft-versus-host disease therapy in some countries [44][45]. Even for allogenic MSCs, however, immunologic rejection has been reported [44].
Muse cells possess unique immunomodulatory properties: allogenic Muse cells can survive and are incorporated into rabbit acute myocardial infarction host tissue for an extended period (>6 months), even without immunosuppressant treatment [6]. In both normal (Wistar) rats and immunocompromised (SCID) mice, intravenously injected human Muse cells survive as neuronal and glial cells in the ischemic brain tissue for 6 months, while MSCs or cells other than Muse cells in MSCs (i.e., SSEA-3(-) non-Muse MSCs that correspond to ~98% of total MSCs) became undetectable in all the tissues in the body within 2 weeks [34][39]. Interestingly, these species-mismatch experiments were conducted without immunosuppressant treatment. Even in non-immunocompromised mice, such as BALB/c mice, intravenously injected human Muse cells homed to damaged glomeruli and survived as glomerular cells for nearly 2 months without immunosuppressant treatment, while non-Muse MSCs became undetectable in the body within 2 weeks [35].
2.7. Bystander Effects of Muse Cells on Tissue Repair
Because Muse cells remain in the host tissue as functional cells for an extended period of time, the anti-inflammatory, anti-apoptotic, and trophic effects brought by Muse cells are long-lasting and effective. Muse cells secrete a variety of factors, including PDGF-A, PDGF-BB, EGF, HGF, VEGF, IL-6, KGF, PGE2, ANG1, TGF-β, bFGF, and SDF-1 that promote wound healing and inhibit apoptosis [6][17][35][47][48]. Hepatocyte growth factor (HGF) and vascular endothelial growth factor (VEGF), which are protective against general tissue damage, are suggested to promote tissue repair in acute myocardial infarction and liver damage models treated with Muse cells [6][48]. In a rat extra-small partial liver transplantation model, HGF and VEGF, which are more highly expressed in Muse cells than in non-Muse MSCs, had a statistically meaningful protective effect on liver sinusoidal endothelial cells [48]. In addition, in a swine hepatectomy model, animals that received allogenic Muse cells exhibited less necrosis, compared with animals that received allogenic MSCs [16]. In damaged brain tissue, Muse cells ameliorated the effects of excitotoxic brain glutamatergic metabolites and suppressed microglial activation, as shown by magnetic resonance spectroscopy and positron tomography, respectively [39]. Thus, Muse cells have greater tissue protective effects than MSCs.
With regard to their anti-inflammatory effect, Muse cells actively produce interleukin-10, transforming growth factor-β, and prostaglandin E2 [17][47]. Granulocyte colony stimulating factor production was recently demonstrated to play a central role in the activities of Muse cells to protect the blood–brain barrier and neural cells in STEC-associated encephalopathy [37].
Muse cells can also produce matrix metalloproteases-1 (MMP1), MMP2, and MMP9. Notably, MMP9 is only produced by Muse cells and not by non-Muse MSCs [6]. MMPs are important for suppressing fibrosis, because they degrade the extracellular matrix. In studies performed using animal models of liver damage, chronic kidney disease, and acute myocardial infarction, intravenous injection of Muse cells provided a statistically meaningful reduction of fibrosis, compared with the MSC/non-Muse MSCs and vehicle groups [9][16][27][40][49].
As mentioned above, Muse cells produce factors that promote neovascularization, represented by VEGF and HGF [6][48]. Several reports, however, indicate that infused Muse cells directly participate in neovascularization by spontaneously differentiating into vascular cells after homing to damaged tissues, such as the post-infarct heart, damaged glomeruli, and damaged liver [6][35][48]. More directly, intravenously injected human Muse cells differentiated into vascular components, CD31+ endothelial cells in the intimal layer and smooth muscle cells in medial layer in a mouse aortic aneurism model [42]. Thus, Muse cells are efficient in vascular protection, as well as in neovascularization.
3. Comparison of the Reparative Effects of Muse Cells and MSCs
The outcomes of tissue repair, functional recovery, anti-apoptotic, anti-inflammatory, and anti-fibrosis effects are all consistently superior in animals injected with Muse cells (Muse group), compared to animals injected with MSC/non-Muse MSCs (MSC/non-Muse MSC group) in disease models. In a rabbit acute myocardial infarction model, for example, the infarct size reduction was ~2.5-fold greater in the Muse group than in the MSC/non-Muse MSC group both at 2 weeks and 2 months [6]. Functional recovery in rat/mouse stroke and hypoxic ischemic encephalopathy models also exhibited statistically meaningful superiority of the Muse group over the MSC/non-Muse MSC group (
p
< 0.001 and
p < 0.01) in the modified neurologic severity, rotarod, and other neurologic function scores at 3 months or even beyond 3 months after administration [38][39]. Statistically significant anti-apoptotic, anti-inflammatory, and anti-fibrosis effects were observed in the Muse cell group, compared with the MSC/non-Muse MSC group in models of mouse chronic kidney disease, mouse hepatitis, and rat lung ischemic-reperfusion [27][35][47]. These differences between Muse cells and MSCs are considered to arise from the differences in their specific homing abilities, the length of survival in the host tissue after intravenous injection, differentiation potential, and immunomodulation. Unlike Muse cells, MSCs do not home to damaged tissue, nor do they remain in the tissue or the body for more than 2 weeks after administration [6][27][35][37][39]. MSCs differentiate into osteocytes, adipocytes, and chondrocytes with lower efficiency than Muse cells and are unable to differentiate into other mesodermal cells or into ectodermal or endodermal lineage cells [26].
< 0.01) in the modified neurologic severity, rotarod, and other neurologic function scores at 3 months or even beyond 3 months after administration [38,39]. Statistically significant anti-apoptotic, anti-inflammatory, and anti-fibrosis effects were observed in the Muse cell group, compared with the MSC/non-Muse MSC group in models of mouse chronic kidney disease, mouse hepatitis, and rat lung ischemic-reperfusion [27,35,47]. These differences between Muse cells and MSCs are considered to arise from the differences in their specific homing abilities, the length of survival in the host tissue after intravenous injection, differentiation potential, and immunomodulation. Unlike Muse cells, MSCs do not home to damaged tissue, nor do they remain in the tissue or the body for more than 2 weeks after administration [6,27,35,37,39]. MSCs differentiate into osteocytes, adipocytes, and chondrocytes with lower efficiency than Muse cells and are unable to differentiate into other mesodermal cells or into ectodermal or endodermal lineage cells [26].
Comparison with Other Stem Cells
Muse cells are distinct from other pluripotent stem cells, such as ES/iPS cells [22], as well as from other somatic stem cells claimed to be pluripotent, such as very small embryonic-like (VSEL) stem cells [50] and multipotent adult progenitor cells (MAPCs) [51], in terms of their proliferative activity, morphology, marker expression, and tissue distribution. Muse cells express pluripotent markers, as mentioned above, and exhibit triploblastic differentiation ability and self-renewability at the single cell level. Compared with ES/iPS cells, however, Muse cells exhibit moderate pluripotency gene expression and are non-tumorigenic; Nanog, Sox2, and Oct3/4 expression levels are lower in Muse cells than in ES/iPSCs, but higher in Muse cells than in general somatic cells, such as fibroblasts [22]. Observations regarding the methylation of the Nanog and Oct3/4 promoter regions support this; these promotor regions are less methylated in Muse cells than in general fibroblasts, whereas those in iPS cells are fully demethylated [18]. On the other hand, expression of genes relevant to the cell cycle is lower in Muse cells than in ES/iPS cells, consistent with the fact that Muse cells exhibit pluripotency but are non-tumorigenic [22]. In relation with this, telomerase activity, an indicator of tumorigenic proliferation, is the same between Muse cells and somatic cells but substantially lower in Muse cells than in iPSCs [22]. The proliferation speed of Muse cells is ~1.3 days/cell division, and expansion is stable until they reach the Hayflick limit; thus, while growth continues on a clinically relevant scale, Muse cells do not proliferate exponentially, unlike ES/iPS cells [1]. Indeed, Muse cells transplanted into the testes of immunodeficient mice did not generate any tumors for up to 6 months [22][30].
Muse cells are distinct from other pluripotent stem cells, such as ES/iPS cells [22], as well as from other somatic stem cells claimed to be pluripotent, such as very small embryonic-like (VSEL) stem cells [50] and multipotent adult progenitor cells (MAPCs) [51], in terms of their proliferative activity, morphology, marker expression, and tissue distribution. Muse cells express pluripotent markers, as mentioned above, and exhibit triploblastic differentiation ability and self-renewability at the single cell level. Compared with ES/iPS cells, however, Muse cells exhibit moderate pluripotency gene expression and are non-tumorigenic; Nanog, Sox2, and Oct3/4 expression levels are lower in Muse cells than in ES/iPSCs, but higher in Muse cells than in general somatic cells, such as fibroblasts [22]. Observations regarding the methylation of the Nanog and Oct3/4 promoter regions support this; these promotor regions are less methylated in Muse cells than in general fibroblasts, whereas those in iPS cells are fully demethylated [18]. On the other hand, expression of genes relevant to the cell cycle is lower in Muse cells than in ES/iPS cells, consistent with the fact that Muse cells exhibit pluripotency but are non-tumorigenic [22]. In relation with this, telomerase activity, an indicator of tumorigenic proliferation, is the same between Muse cells and somatic cells but substantially lower in Muse cells than in iPSCs [22]. The proliferation speed of Muse cells is ~1.3 days/cell division, and expansion is stable until they reach the Hayflick limit; thus, while growth continues on a clinically relevant scale, Muse cells do not proliferate exponentially, unlike ES/iPS cells [1]. Indeed, Muse cells transplanted into the testes of immunodeficient mice did not generate any tumors for up to 6 months [22,30].
Pluripotent stem cells were recently classified into two discrete states, ‘naïve’ and ‘primed’, based on their growth factor dependency, intracellular signaling, marker expression, and differentiation potential [52]. For example, naïve pluripotent stem cells depend on the LIF/STAT3 and BMP4 pathways or LIF + small molecule inhibitors (called 2i) to sustain their self-renewability [53], whereas primed pluripotent stem cells depend mainly on FGF and activin/transforming growth factor β to stably promote their self-renewal [54]. Muse cell properties are more similar to those of primed pluripotent stem cells than naïve pluripotent stem cells, such as ES/iPS cells, because Muse cells do not require FGF to maintain their proliferation and self-renewal abilities.
VSELs (3–5 μm; smaller than red blood cells) found in the peripheral blood, umbilical cord blood, and reproductive tissues exhibit positivity for Sca1, CD34, CXCR4, and SSEA-1 and negativity for Lin and CD45 [50]. In contrast to VSELs, Muse cells are found not only in the bone marrow and peripheral blood but also in organ connective tissue. Human Muse cells in the bone marrow and organs are 13~15 μm, and those in the peripheral blood are ~10 μm; thus, Muse cells are considerably larger than VSELs [1][4]. Marker expression also differs between Muse cells and VSELs; Muse cells from the bone marrow and organs are double-positive for SSEA-3 and CD105, an MSC marker, whereas peripheral blood-derived Muse cells are consistently double-positive for SSEA-3 and CD45, a white blood cell marker. The expression of CD45 differs between Muse cells and VSELs. In addition, VSELs are positive for CD34, whereas Muse cells are not [1][4].
VSELs (3–5 μm; smaller than red blood cells) found in the peripheral blood, umbilical cord blood, and reproductive tissues exhibit positivity for Sca1, CD34, CXCR4, and SSEA-1 and negativity for Lin and CD45 [50]. In contrast to VSELs, Muse cells are found not only in the bone marrow and peripheral blood but also in organ connective tissue. Human Muse cells in the bone marrow and organs are 13~15 μm, and those in the peripheral blood are ~10 μm; thus, Muse cells are considerably larger than VSELs [1,4]. Marker expression also differs between Muse cells and VSELs; Muse cells from the bone marrow and organs are double-positive for SSEA-3 and CD105, an MSC marker, whereas peripheral blood-derived Muse cells are consistently double-positive for SSEA-3 and CD45, a white blood cell marker. The expression of CD45 differs between Muse cells and VSELs. In addition, VSELs are positive for CD34, whereas Muse cells are not [1,4].
MAPCs (8~10 μm), which locate in the bone marrow, are positive for markers related to MSCs (CD13, CD44, CD73, CD90, and CD105) and negative for hematopoietic (CD34, CD45, and CD117) and endothelial (CD34 and CD309) markers [51]. Bone marrow- and organ-derived Muse cells express MSC markers, such as CD29, CD90, and CD105, together with SSEA-3 [22], whereas peripheral blood-Muse cells express CD45 and SSEA-3 [4].
Together, these findings clearly demonstrate that Muse cells are distinct from ES/iPS cells, VSELs, and MAPCs.