Neurodegenerative diseases (NDs) including Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and Huntington’s disease are incurable and affect millions of people worldwide. The development of treatments for this unmet clinical need is a major global research challenge. Computer-aided drug design (CADD) methods minimize the huge number of ligands that could be screened in biological assays, reducing the cost, time, and effort required to develop new drugs.
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
Neurodegenerative diseases (NDs) are incurable and debilitating conditions that result in progressive degeneration and/or death of nerve cells in the central nervous system (CNS) [1][2][3]. Dementia rates are alarmingly on the rise worldwide. There are over 50 million people worldwide living with dementia in 2020, with nearly 60% living in low- and middle-income countries [4]. This number will almost double every 20 years, reaching 82 million in 2030 and 152 million in 2050 [4]. The number of people with dementia in the UK is predicted to be around 1.14 million by 2025 and 2.1 million by 2051, an increase of 40% over the next 5 years and 157% over the next 31 years [5].
Neurodegenerative diseases (NDs) are incurable and debilitating conditions that result in progressive degeneration and/or death of nerve cells in the central nervous system (CNS) [1,2,3]. Dementia rates are alarmingly on the rise worldwide. There are over 50 million people worldwide living with dementia in 2020, with nearly 60% living in low- and middle-income countries [4]. This number will almost double every 20 years, reaching 82 million in 2030 and 152 million in 2050 [4]. The number of people with dementia in the UK is predicted to be around 1.14 million by 2025 and 2.1 million by 2051, an increase of 40% over the next 5 years and 157% over the next 31 years [5].
The UK Prime Minister’s Challenge on Dementia was launched in 2015 to identify strategies to tackle dementia by 2025 [6]. Current therapies for NDs treat symptoms, not the underlying pathological changes. There is a clear and unmet clinical need to develop new therapies based on understanding the molecular pathologies. One of the most promising approaches is to develop novel therapeutics using computer-aided drug design (CADD) [7][8].
The UK Prime Minister’s Challenge on Dementia was launched in 2015 to identify strategies to tackle dementia by 2025 [6]. Current therapies for NDs treat symptoms, not the underlying pathological changes. There is a clear and unmet clinical need to develop new therapies based on understanding the molecular pathologies. One of the most promising approaches is to develop novel therapeutics using computer-aided drug design (CADD) [7,8].
2. Computer-Aided Drug Design
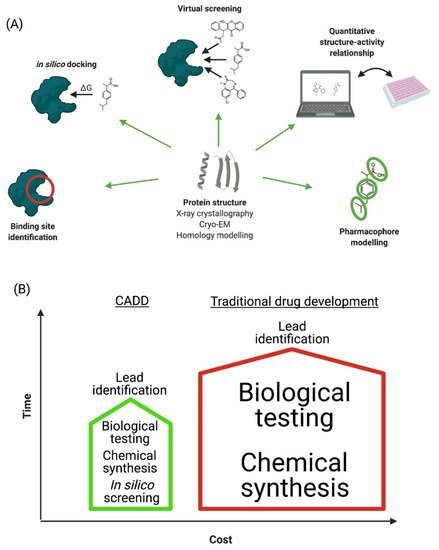
“Computer-aided drug design” (CADD) refers to the application of computational modelling approaches to drug discovery. Drug discovery is an expensive and time-consuming process with the average approved drug requiring 10 to 15 years to develop with an estimated cost of 0.8–2 billion USD [9]. Many licensed drugs, such as captopril, dorzolamide, oseltamivir, aliskiren, and nolatrexed, were all optimized using CADD [10], and a large number of publications describe the successful design and discovery of leads/drugs using CADD [11][12][13]. The major steps involved in CADD are summarized in
“Computer-aided drug design” (CADD) refers to the application of computational modelling approaches to drug discovery. Drug discovery is an expensive and time-consuming process with the average approved drug requiring 10 to 15 years to develop with an estimated cost of 0.8–2 billion USD [9]. Many licensed drugs, such as captopril, dorzolamide, oseltamivir, aliskiren, and nolatrexed, were all optimized using CADD [10], and a large number of publications describe the successful design and discovery of leads/drugs using CADD [11,12,13]. The major steps involved in CADD are summarized in A and discussed in the following sections. The main goal of CADD is to reduce these timescales and costs without affecting quality (
B) [14]. Importantly, CADD can be used in most stages of drug development: from target identification to target validation, from lead discovery to optimization, and in preclinical studies. It is therefore estimated that CADD could reduce the cost of drug development by up to 50% [15][16].
B) [14]. Importantly, CADD can be used in most stages of drug development: from target identification to target validation, from lead discovery to optimization, and in preclinical studies. It is therefore estimated that CADD could reduce the cost of drug development by up to 50% [15,16].
Figure 1.
(
A
) Schematic representation of CADD process. (
B
) Comparison of traditional and computer-aided drug development in terms of time and cost investments.
2.1. Drug Target Selection
Drug target selection is the first step of structure-based drug design. This involves identifying and determining the structures of the relevant proteins [17]. Understanding and characterization of the molecular biology of the targeted disease are therefore necessary before the initiation of any active compound search process.
2.2. Determination of the Protein Structure
An in-depth understanding of biological processes is still often hampered by a lack of detailed knowledge of protein structures [18]. The determination of the structure of the target protein is a prerequisite for CADD [19]. Structural elucidation of the target protein can be performed by experimental tools including, but not limited to, nuclear magnetic resonance (NMR) spectroscopy, Cryo-EM, and X-ray crystallography [20][21].
An in-depth understanding of biological processes is still often hampered by a lack of detailed knowledge of protein structures [18]. The determination of the structure of the target protein is a prerequisite for CADD [19]. Structural elucidation of the target protein can be performed by experimental tools including, but not limited to, nuclear magnetic resonance (NMR) spectroscopy, Cryo-EM, and X-ray crystallography [20,21].
2.3. Homology Modelling
Despite the current revolution in structural studies, in particular the recent developments in cryo-EM, the detailed structures of a large number of proteins, and especially membrane proteins (which are over-represented amongst drug targets), have not been determined [18][22]. Homology modelling is an approach to estimate the structure of a target protein based on structural data from proteins with sequence homology to the target [23].
Despite the current revolution in structural studies, in particular the recent developments in cryo-EM, the detailed structures of a large number of proteins, and especially membrane proteins (which are over-represented amongst drug targets), have not been determined [18,22]. Homology modelling is an approach to estimate the structure of a target protein based on structural data from proteins with sequence homology to the target [23].
For instance, a homology model of human catechol-O-methyltransferase (COMT) was constructed utilizing the X-ray crystal structure of rat COMT to design anti-PD drugs by performing ligand docking, resulting in the discovery of nine putative inhibitors. Another example involves a cysteine protease from
Xanthomonas campestris
(an aerobic, Gram-negative rod-shaped bacterium known to cause black rot in crucifers by darkening the vascular tissues). The active site of this enzyme is homologous to human cathepsin B enzyme (hCB), the activity of which contributes to the reduction of the amyloid peptide by proteolytic cleavage of Aβ1-42, offering a protective role against AD [24].
2.4. Identification of Binding Sites
When the three-dimensional structure of the target protein is determined, the next step is the identification of potential binding sites for small molecules. This process can be conducted using various algorithms for computing and identifying binding pockets [25][26][27].
When the three-dimensional structure of the target protein is determined, the next step is the identification of potential binding sites for small molecules. This process can be conducted using various algorithms for computing and identifying binding pockets [25,26,27].
2.5. Molecular Dynamics Simulation
Molecular dynamics (MD) simulations are a theoretical tool to discover the configurations and dynamic behaviours of molecules, providing atomic-level insight into drug mechanisms of action [13]. MD may also help to reveal the aggregation pathway of neurotoxic protein aggregates and thus aid in the design of new inhibitors [28].
2.6. Molecular Docking Studies
Molecular docking is a computational procedure that predicts the lowest energy binding conformations of one molecule to a second (usually a small drug-like molecule to a protein). Accordingly, molecular docking procedures, along with their different scoring systems, are frequently utilized to predict the binding modes and affinities between chemical compounds and drug binding sites on biological macromolecules [29][30].
Molecular docking is a computational procedure that predicts the lowest energy binding conformations of one molecule to a second (usually a small drug-like molecule to a protein). Accordingly, molecular docking procedures, along with their different scoring systems, are frequently utilized to predict the binding modes and affinities between chemical compounds and drug binding sites on biological macromolecules [29,30].
2.7. Virtual Screening
Virtual screening (VS) is the process of screening small molecule libraries in silico to identify chemical structures that may bind to a drug target [31][32][33].
Virtual screening (VS) is the process of screening small molecule libraries in silico to identify chemical structures that may bind to a drug target [31,32,33].
2.8. Quantitative Structure—Activity Relationship Study
Quantitative structure—activity relationship (QSAR) methods are conducted to correlate a biological response (e.g., enzyme activity, cell viability, etc.) to the chemical properties of a set of molecules [34][35][36].
Quantitative structure—activity relationship (QSAR) methods are conducted to correlate a biological response (e.g., enzyme activity, cell viability, etc.) to the chemical properties of a set of molecules [34,35,36].
2.9. Pharmacophore Modelling
Pharmacophore modelling deals with finding the optimal shapes and charge distributions for binding of a small molecule to a biological macromolecule. Pharmacophore modelling is commonly implemented to rapidly specify potential lead compounds [37][38].
Pharmacophore modelling deals with finding the optimal shapes and charge distributions for binding of a small molecule to a biological macromolecule. Pharmacophore modelling is commonly implemented to rapidly specify potential lead compounds [37,38].
3. A Roadmap for Implementing CADD in ND Drug Design
Even with the number of successful implementations of CADD in modern drug discovery, it has its limitations. Molecules designed in silico utilizing computational and theoretical chemistry sometimes do not work in real biological systems [39][40]. In general, poor pharmacokinetics and/or pharmacodynamics result in only 40% of drug candidates passing phase I clinical trials [41]. Moreover, each computational technique depends on pre-determined algorithms that have their own limitations. CADD results must be validated in real biological systems, as many molecules that appear to bind in silico do not show the desired activity in vitro. Another limitation of CADD is that all tools for designing and discovery of new drugs are based on algorithms that, by necessity, simplify the underlying physics and chemistry and, therefore, have a variety of limitations that necessitate the continuous updating of these algorithms to enhance the accuracy and thus the provision of new drugs [42][43][44][45][46][47][48]. Furthermore, the shortage of experimental data regarding predicted absorption, distribution, metabolism, excretion, and toxicity results has led to several published failures [49][50][51][52][53].
Even with the number of successful implementations of CADD in modern drug discovery, it has its limitations. Molecules designed in silico utilizing computational and theoretical chemistry sometimes do not work in real biological systems [159,160]. In general, poor pharmacokinetics and/or pharmacodynamics result in only 40% of drug candidates passing phase I clinical trials [161]. Moreover, each computational technique depends on pre-determined algorithms that have their own limitations. CADD results must be validated in real biological systems, as many molecules that appear to bind in silico do not show the desired activity in vitro. Another limitation of CADD is that all tools for designing and discovery of new drugs are based on algorithms that, by necessity, simplify the underlying physics and chemistry and, therefore, have a variety of limitations that necessitate the continuous updating of these algorithms to enhance the accuracy and thus the provision of new drugs [162,163,164,165,166,167,168]. Furthermore, the shortage of experimental data regarding predicted absorption, distribution, metabolism, excretion, and toxicity results has led to several published failures [169,170,171,172,173].
To overcome the limitations and improve the accuracy of CADD it is necessary to update and develop software and associated algorithms, validate with experimental data, use reliable databases (e.g., PDB), and use algorithms that give docking scores that accurately predict in vitro binding with comprehensive and fully retrospective coverage of the published literature [54][55][56]. For example, by September 2020, the Cambridge Structural Dataset (CSD) acquired more than 1.8 million entries, which may help with future developments in small molecule structural modelling [57]. Consequently, the above-mentioned tools could help with future design of pharmacophores that possess the desired biological activity [58][59][60].
To overcome the limitations and improve the accuracy of CADD it is necessary to update and develop software and associated algorithms, validate with experimental data, use reliable databases (e.g., PDB), and use algorithms that give docking scores that accurately predict in vitro binding with comprehensive and fully retrospective coverage of the published literature [174,175,176]. For example, by September 2020, the Cambridge Structural Dataset (CSD) acquired more than 1.8 million entries, which may help with future developments in small molecule structural modelling [177]. Consequently, the above-mentioned tools could help with future design of pharmacophores that possess the desired biological activity [178,179,180].
One of the main reasons for implementing in silico drug design is to predict the ligand–target binding in terms of binding site and binding strength. To predict potential ligands to treat NDs, novel target proteins must be identified and studied, and the resulting docking studies should be validated in vitro and eventually in the clinic [61][62][63].
One of the main reasons for implementing in silico drug design is to predict the ligand–target binding in terms of binding site and binding strength. To predict potential ligands to treat NDs, novel target proteins must be identified and studied, and the resulting docking studies should be validated in vitro and eventually in the clinic [181,182,183].
In the meantime, there is no effective treatment to cure NDs, although many treatments are available that offer minor improvement of symptoms [2]. The development of effective treatments is further hindered by the BBB that excludes many molecules from the CNS parenchyma [64][65][66]. Accordingly, clinical effectiveness of a potential drug is not guaranteed even with positive data in silico, in vitro, and in vivo [67][68][69][70].
In the meantime, there is no effective treatment to cure NDs, although many treatments are available that offer minor improvement of symptoms [2]. The development of effective treatments is further hindered by the BBB that excludes many molecules from the CNS parenchyma [184,185,186]. Accordingly, clinical effectiveness of a potential drug is not guaranteed even with positive data in silico, in vitro, and in vivo [187,188,189,190].
New experimental approaches including genome-wide association studies (GWAS) [68][71][72], CRISPR-Cas9 technology [73][74][75], high throughput screening (HTS) [76], organ-on-chip technologies [77][78], functional MRI (fMRI) techniques [79][80], and positron emission tomography (PET) [81] may lead to new drug targets for NDs, which can feed into future CADD projects.
New experimental approaches including genome-wide association studies (GWAS) [188,191,192], CRISPR-Cas9 technology [193,194,195], high throughput screening (HTS) [196], organ-on-chip technologies [197,198], functional MRI (fMRI) techniques [199,200], and positron emission tomography (PET) [201] may lead to new drug targets for NDs, which can feed into future CADD projects.
Being incurable, the NDs are major challenges to healthcare providers and research scientists. The accelerating increase in the numbers of affected people adds more impetus to tackle NDs. Developing a better understanding of NDs and the underlying molecular pathophysiology will provide more opportunities to develop novel treatments in the near future. This may be achieved with the incorporation of computational tools. CADD can have a major impact on drug discovery by saving both time and money and reducing the risk of following up with the development of non-viable leads.