Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Lily Guo and Version 1 by Natalia Rivera-Torres.
DNA repair mechanisms include nucleotide pool sanitization (nucleotide pool sanitization), direct repair (DR), base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), homologous recombination repair ( HRR) and non-homologous end joining (NHEJ).
The mechanism of action of ssODN-directed gene editing has been a topic of discussion within the field of CRISPR gene editing since its inception. Multiple comparable, but distinct, pathways have been discovered for DNA repair both with and without a repair template oligonucleotide.
- CRISPR/Cas
- homology directed repair
- DNA repair
1. Introduction
The mechanism of action and regulation of ssODN-directed gene editing has been a topic of discussion and of great interest for over 20 years. With the rapid development and ease-of-use issue of the latest gene editing technology, CRISPR/Cas has permitted the elucidation and further characterization of the gene editing mechanisms. The CRISPR/Cas system functions naturally in the adaptive immunity pathway of bacteria. Through repurposing, CRISPR/Cas has emerged as the preferred system to catalyze site-specific DNA cleavage [1,2,3,4,5,6][1][2][3][4][5][6]. Cas assembles with a designed gRNA to create a complex that can cut at any desired site in a chromosome. A protospacer adjacent motif (PAM) is required for Cas9 binding, and the target must be upstream of a 5′-NGG-3′ site (with SpCas9) or downstream of 5′-TTTN-3′ (with AsCas12a). The Cas/gRNA complex scans the genome for the PAM sequence. When the Cas/gRNA binds to a PAM, it then locally unwinds the DNA adjacent to the PAM (upstream with Cas9, downstream with Cas12a); if the target region of the gRNA can bind with a perfect match, the Cas nuclease then cuts both strands of DNA [7,8][7][8]. Once the CRISPR/Cas complex has created a DSB, cells employ several pathways to repair the damage; these pathways include nonhomologous end joining (NHEJ), microhomology-mediated end joining (MMEJ), and homology-directed repair (HDR) [7,9,10][7][9][10]. NHEJ can occur through canonical NHEJ (C-NHEJ), which ligates the broken ends back together. There is an alternative end joining pathway (alt-NHEJ), in which one strand of the DNA on either side of the break is resected to repair the lesion [10,11][10][11]. In traditional DNA repair, NHEJ typically leads to scarless repair, where the DNA ends are religated without losing any sequence information. In the presence of CRISPR constructs, however, this scarless repair leads to recapitulation of the gRNA recognition site, causing recleaving, which shifts the overall repair outcome likelihood significantly in favor of sequence-changing repair outcomes [12]. MMEJ can be utilized to repair broken DNA ends by utilizing patches of microhomology on either end of the break site, leading to specific deletions upon repair [13,14,15,16][13][14][15][16]. These repair methods are error-prone, meaning that the lesion is repaired imperfectly, resulting in insertions or deletions (indels). If there is a DNA molecule within the local vicinity of the break that shares homology with the region around the double-strand break, then the homologous DNA can be utilized as a template to repair the break via the homology-directed repair (HDR) pathway. Utilization of various CRISPR/Cas systems have greatly increased the pace and lowered the cost of research, expanding the possibilities and allowing for the genetic manipulation of cells and organisms that have historically been difficult to modify [17]. A major challenge in this area is changing a DNA sequence to define gene function through HDR, rather than to delete the gene function via NHEJ/MMEJ, which results in total dysfunction [18,19][18][19].
The correction of single base mutations in cells has long been the ultimate goal of gene editing with single stranded DNA oligonucleotides (ssODN) [20,21,22][20][21][22] and its mechanism and regulation has been a greatly discussed topic over the last 15 years [21,22,23,24,25,26,27,28,29,30,31][21][22][23][24][25][26][27][28][29][30][31]. With these attempts, a number of elaborate models based on Homology Directed Repair (HDR) have been established [32,33,34][32][33][34]. With the incorporation of CRISPR/Cas9 to the gene editing toolbox, many paired this new technology with ssODNs, leading to higher efficiency of the precise repair of point mutations. By reengineering key reaction components, such as the interaction between CRISPR/Cas9 and the PAM sites [35], chemically modifying the Cas9 [36], ssODN [37], and sgRNA [38] HDR accuracy and efficiency has been significantly increased.
In 2017, Rivera-Torres et al. [39] introduced the Excision and Corrective Therapy (ExACT) model. This model proposed an overall mechanism by which short oligonucleotides and a CRISPR/Cas9 RNP execute the repair of a point mutation in which the donor DNA acts as a replication template for the repair of the mutant base. In comparison to previously published models [33[33][40],40], the original ExACT model was much simpler and was based on the well-accepted and standard model of Double-Strand Break Repair [35,41,42][35][41][42]. In their work, Rivera-Torres et al. demonstrated that it is possible to direct single nucleotide exchange in an efficient manner, but also found indels accompany single base repair. This general concept, outlined in Figure 1, explains the overall reaction of how single-stranded oligonucleotides direct point mutation repair in mammalian cells [22,27,43,44][22][27][43][44]. The double stranded break in the chromosome induced by CRISPR/Cas activity produces two exposed DNA ends both upstream and downstream of the cut site. These ends are subject to 5′ > 3′ resection, as seen in NHEJ/MMEJ, but these mechanisms are independent of the template-driven repair utilized by ExACT. In the described ExACT model, an ssODN template interrogates both ends of the cut site simultaneously, bridging the gap between ends. This allows for DNA synthesis across the gap, with synthesis occurring in a 5′ to 3′ direction, ending when DNA synthesis catches up to the end of the resected DNA end, leading to dissociation of the synthesis complex and ligation of the newly-synthesized strand into the subsequent DNA strand. After this step of repair, the oligonucleotide is no longer necessary, and must dissociate from the complex in order for the repair to be completed. Once the repair template ssODN is dissociated, the newly-synthesized stretch of DNA on the opposite strand can be used as a template for synthesis-driven fill-in of the remaining bases, followed by varying degrees of resection of the broken ends and extension by the DNA replication machinery. These steps are independent of the desired or intended repair process, HDR or NHEJ. The gap created by the original double-strand break is repaired through the utilization of an exogenous piece of DNA that serves as a source of genetic information and the template for replication activity and gap filling.
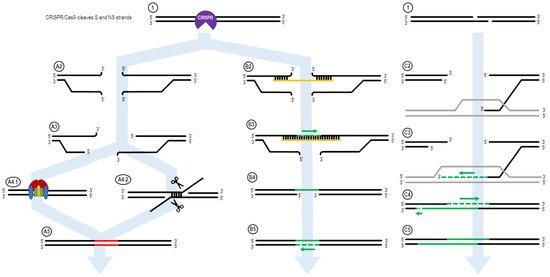
Figure 1. The ExACT DNA repair pathway, as outlined by Rivera-Torres et al. Upon CRISPR-mediated cleavage or breakage of the DNA (1), several possible pathways may be undertaken in order to facilitate DNA repair. In the absence of an ssODN oligo, or if the ssODN is not in the local vicinity of the broken DNA ends, the non-oligo repair pathway (A2–A5) is used. The CRISPR-Cas complex dissociated from the DNA ends (A2), and resection of the DNA ends may occur (A3). Then, either nonhomologous end joining (NHEJ, A4.1) or microhomology-mediated end joining (MMEJ, A4.2) occurs to repair the broken DNA ends. This results in an indel in the resulting DNA product, as bases are added or deleted via NHEJ and MMEJ. In the presence of an oligo, the ExACT pathway is undertaken (B2–B5). The oligo bridges both sides of the broken DNA ends (B2) and facilitates synthesis-driven repair on one strand (B3). The oligo then dissociated from the repaired DNA strand (B4), so the repaired strand can in turn be used as a repair template for a second synthesis-driven repair event on the other DNA strand (B5). This results in repair containing added, deleted or altered sequence information from the repair template ssODN. In the presence of a homologous gene, synthesis-dependent strand annealing can occur. One 3′-end on either end of the DNA break interrogates the homologous gene and forms a D-loop (C2). The 3′-end is extended via DNA synthesis, expanding the D-loop (C3). After a period of extension, the newly-lengthened 3′-end dissociates from the homologous gene, and binds to its complementary sequence on the other end of the DNA break—bridging the gap (C4). Further DNA synthesis fills in the remaining bases according to their respective complementary sequences (C5).
The ExACT pathway, in essence, shows some similarity to the synthesis-dependent strand annealing (SDSA) mechanism of DNA repair [44,45,46][44][45][46]. In SDSA, a DNA break is subjected to 5′-to-3′ resection on both ends of the break site, as is seen in MMEJ. In the presence of a homologous gene from a sister chromosome, however, one single-stranded 3′ end of the broken DNA interrogates the sister gene, initiating DNA synthesis and elongation of the 3′ end in accordance with the sequence of the sister gene. Once extended far enough to bridge the DNA break gap, the elongated 3′ end dissociates from the sister gene and re-binds to the opposite end of the break site, allowing the resulting complex to be filled in through DNA synthesis and ligated back into a correctly repaired DNA strand. This differs from the other sister chromosome repair model, the Double-Holliday Junction Model [47], as in SDSA where there are no DNA strands that move from one chromosome to another. In SDSA, there was no crossing-over event seen, while the Double-Holliday Junction Model requires crossing over for complete DNA repair [48]. In common discussions related to CRISPR gene editing, SDSA is invoked as a repair mechanism for oligonucleotide-driven repair. The mechanism through which oligonucleotide-driven repair would occur, however, does not fit the traditional description of SDSA. In SDSA, the resected DNA end of a broken DNA strand interrogates an unbroken dsDNA template from a sister chromosome. In oligonucleotide-driven HDR, however, the oligo interrogates the broken DNA ends instead. This difference, while slight, can lead to multiple different downstream effects that are unique to each mechanism. In SDSA, for instance, incorporation of the repair template directly into the broken DNA is impossible, as no crossing over events occur, leading to ligation between the broken DNA and the repair template.
In relation to CRISPR gene editing repair, the distinction between semi-conservative and conservative DNA repair can be seen when editing members of homologous gene families. In multiple papers by several labs studying CRISPR editing in sickle cell anemia, targeting the hemoglobin beta gene (HBB) with a CRISPR/Cas construct led, in a small number of cases, to a conversion of a segment of the HBB gene to that of hemoglobin delta (HBD), effectively creating a novel HBB/HBD chimeric gene [32,49,50][32][49][50]. When the hemoglobin delta gene was sequenced in these HBB-HBD converted cells, the HBD conversion was found to be non-reciprocated, with no segment of the HBB sequence present within the HBD gene. This points to a fully conservative method of synthesis-directed gene repair and excludes the semi-conservative Double Holliday Junction Model from consideration as a potential pathway utilized to generate the given data. While DHJ remains an established and known pathway for some forms of gene repair and conversion, there remains no evidence of this particular pathway occurring in CRISPR-directed gene editing. Furthermore, when examining repair mechanisms utilized in the presence of a ssODN repair template, DHJ becomes impossible as a repair mechanism, as the short length of the ssODN would result in chromosome truncation if semi-conservative gene repair did take place.
2. ExACT Pathway vs. SDSA DNA Repair Mechanism
Figure 1 shows the ExACT pathway. As explained by Rivera-Torres et al. [39], the ExACT pathway requires, as a prerequisite to repair, CRISPR-cleaved DNA ends and a repair template with a specific genetic change. Upon cleavage by the CRISPR-Cas complex, multiple pathways can occur concurrently and simultaneously at the broken DNA ends. In the absence of a homology donor, or if the donor is not in the local area of the cut DNA ends, nonhomologous end joining (NHEJ) and microhomology-mediated end joining (MMEJ) can occur. These two lossy DNA repair pathways both take advantage of DNA resection at the cut site in order to open the DNA ends for repair. In the presence of Ku70/80 stabilizing the DNA ends (Figure 1, A4.1), NHEJ occurs to seal the DNA ends with minimal sequence loss at the cut site [11,51][11][51]. Indels that occur through NHEJ are typically small (>30 bp) and random; indels across a population of cut DNA ends, both in vitro and in vivo, will have a plurality of indels at the cut site, rather than 1–3 major ones.
In many cases, however, MMEJ is readily seen at a cut site, even with just the slightest microhomology around the cut site. During MMEJ, single-stranded resection of the broken DNA ends reveal patches of DNA both upstream and downstream of the cut site that incidentally share sequence homology. These areas of microhomology associate with each other, leading to temporary stabilization of the broken DNA ends (Figure 1, A4.2). The degree of stabilization is dependent on the extent of the microhomology, but as can be seen in Figure 4D, as little as 4 bp is enough to establish strong enough microhomology to overcome NHEJ as the predominant indel source.
When an ssDNA repair template is present in the reaction, however, a separate pathway is taken. This pathway, displayed on the right side of Figure 1, is the ExACT pathway. The ssDNA repair template bridges the DNA break on one strand and associates with the perfect-match sequences both upstream and downstream of the cut site. Depending on the speed of DNA end resection and the size of the DNA oligo, this process may curtail the resection process by bridging the gap further downstream of any resected bases. Once the cut site is stabilized with the oligonucleotide, the missing bases across the cut site are filled in via DNA polymerase, using the repair oligo as a negative-strand guide. This allows any changes intentionally incorporated into the repair template to be copied into the repaired DNA strand, including base changes, insertions, or deletions (B3). Once the strand complementary to the repair template is fully repaired and ligated, the oligonucleotide dissociates from the junction, leaving a single-strand gap around what was once the CRISPR cleavage site (B4). This single-stranded gap is then filled in by DNA polymerase using the previously repaired DNA sequence as a guide (B5). This results in a cleanly repaired DNA template with no need for incorporation of the ssDNA oligo to facilitate repair.
The mechanism of the ExACT pathway bears many similarities in concept to the Synthesis-Dependent Strand Annealing pathway. Both allow for DNA repair to occur without disruption of the reference DNA sequence, as compared to the Double Holliday Junction Model, which leads to a crossover event between the repair reference DNA sequence and the cut DNA ends. One major mechanistic difference between the two pathways, however, is the simultaneous binding of both open DNA ends in the ExACT model. In SDSA, the repair template, a double-stranded DNA homolog, interrogates a single broken DNA end and allows for polymerization from that end to extend the 3′-end of the cut. This extended 3′-end then bridges the break site, binding to its complement on the opposite end and allowing for the filling in of any gaps in the sequence through polymerase activity [52]. The two-step polymerization process employed in SDSA differs from the single-step bridging of the gap outlined in the originally published ExACT manuscript [39].
This distinction between a single gap-bridging event and a two-step polymerization process is a subtle difference, and further research into the physiochemical structure of these D-loop formations will need to be performed in order to empirically ascertain which mechanism, if either, are being employed in in vivo gene editing reactions. Further testing would then have to be performed in order to verify that the structures formed within the in vitro system replicate those seen in live cells. Regardless, the presence of a one-step or two-step gap-bridging event would be able to be seen in the secondary indels observed in non-perfect HDR events. These events are typically much more rare in gene editing reactions, as they require one of these structures to resolve incorrectly. Due to how rare these events are in in vivo gene editing, it is likely that, even if discovered, these secondary incorrect recombinations would also be found in the appropriate quantities to evidence the necessary events in order to distinguish mechanisms as subtly different as a one-step or two-step gap bridging event taking place. The overall rarity of these events being recordable in in vivo editing, in addition to the massive amount of targeting, clonally expanding, and sequencing that would be necessary to generate such datasets would prevent any conclusive statements about the formation of these atypical repair outcomes. However, using the in vitro system, such specific hypotheses can be tested with ease.
The in vitro system has served to examine the molecular mechanism of CRISPR-mediated gene repair in a tightly-controlled, optimized system [53,54][53][54]. In this system, the CRISPR-Cas RNP complex provides the double-stranded cleavage and a mammalian cell-free extract provides the enzymatic activity to promote activation of the DNA repair pathways in the presence of a donor DNA template. Briefly, a CRISPR cleavage reaction is performed on a plasmid construct containing a gene of interest (in this case, LacZ). The linearized plasmid, cleaved at the CRISPR target site, is then re-circularized using a cell-free extract isolated from live cells, containing all of the necessary components of DNA repair, at the proper concentrations and availabilities. This effectively mimics DNA repair seen in a live cell, without having to account for many of the confounding factors commonly seen in in vivo gene editing, such as nuclear uptake and low DNA cleavage efficiencies [53].
Figure 2 shows the partial outcomes of an experiment that was carried out within the in vitro system. The goal of the experiment was to test single base repair competencies using an ssODNs. Due to the highly optimized nature of these reactions in vitro, we were only able to screen sequences that showed editing through the bacterial LacZ readout [53].
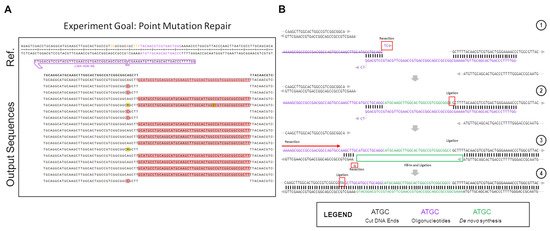
Figure 2. Atypical repair outcomes caused by ssODN dimerization in in vitro gene editing. The partial outcome data of one gene editing reaction, carried out using Cas12a in the in vitro system, are shown here. (A) The experimental results. The LacZ gene was repaired with a semi-symmetrical oligo, with one 40 bp and one 29 bp homology arm, with a single base repair (A > C) in the middle. (B) Depiction of the pathway of repair that led to the 48 bp insertions seen in the experimental output. (1) The dimerized ssODN (purple) associated with the cut DNA ends (black) in order to facilitate the generation of the products seen. (2) The dimerized ssODN created a makeshift gap that allowed for DNA synthesis to fill in according to the template (green). Ligation occurs after the green bases are synthesized, incorporating the actual oligo into the top strand of the downstream end as a long 5′-overhang. (3) After ligation, the unincorporated ssODN dissociates from the complex, and the newly-extended 5′-overhang associates with a 5bp stretch of microhomology on the upstream end of the cut site. The 5′-end upstream of this microhomology patch is resected or cleaved, as well as the single mismatched base on the exposed 5′-end of the bottom strand. (4) Ligation occurs after resection on the top strand, leading to a complete top strand. The bottom strand, meanwhile, is filled in via synthesis, using the top strand as a template.
A significant portion of the non-perfect repair outcomes in Figure 2A consist of 48 bp insertions of the same DNA sequence. This sequence is consistent with the repair oligo, but in the opposite orientation, as would be expected if the oligo was undergoing normal DNA repair. Instead, what appears to be happening in this case is an in-line incorporation of the oligo against its normal orientation, with the negative sense-strand oligo being incorporated in-line with the sense-strand gene sequence. The high rate at which this specific indel was seen in the outcome points to a highly-reproducible event being the culprit for the peculiar recombination. While accounting for many different potential causes for the indel, most, while still possible, lacked the unique determined repair outcome, or implied the possibility of several varying outcomes, which were not seen in the results, and as such were eliminated from the contention as the cause for these repairs. Upon full examination of the potential causes for such a replicable, but atypical insertion, it was eventually realized that an ssODN dimerization had occurred, as displayed in step 1 of Figure 2B. Using Integrated DNA Technologies OligoAnalyzer tool, the delta-G of the dimerized oligo was found to be −16.38 Kcal/mol, with 8 bp of homology on the 3′-ends of the ssODN. There was another product with a more energetically favorable delta-G, at −22.78 kCal/mol, but this dimer created 3′ overhangs, which would prevent the dimer from associating with the free DNA ends left after Cas12a cleavage (Figure S1).
This atypical repair outcome provides an interesting example of potential DNA repair after CRISPR-Cas cleavage, as in this situation, it appears as if both ExACT repair and SDSA-like repair is occurring sequentially within these repair events. The proposed pathway for the atypical repair is seen in Figure 2B. First, the oligo associates with the downstream DNA end, while simultaneously dimerizing with itself at the previously described 8bp stretch. This complex is incredibly similar to the one-step gap-bridging event outlined in the ExACT pathway, but with a dimerized oligo being used in place of the top strand of the DNA upstream of the cut site. The final two bases of the ssODN, which do not match complimentarily, are removed via flap endonucleases or the limited 3′>5′ resection seen in DSB repair complexes. Using the properly oriented ssODN as a template, the gap is then filled in via DNA synthesis, with the end ligated to the downstream plasmid DNA end. Once this ligation occurs, the properly oriented oligonucleotide is not necessary for further repair, and in fact may hinder further repair if it remains bound.
This resulting repair outcome, however, still does not lead to recircularization of the plasmid, which had to have occurred for these DNA sequences to be amplified. This repair outcome alone would have merely led to an extended linearized plasmid, which would not have been able to be amplified with the utilized primers. Thus, it was determined that, after this initial repair event, a second synthesis-driven repair would have had to take place in order to properly re-circularize the plasmid. Due to the incorporation of the improperly-oriented oligo due to dimerization, the DNA ends at the cut site are changed, with the downstream end now containing a long 5′ overhang that happens to match a 5 bp microhomology patch upstream of the cut site. This complex, displayed in Step 3 of Figure 2B, aligns more closely with the repair complex seen in the SDSA-like repair. There are still discrepancies as this complex is formed with a long 5′ overhang, as opposed to a long 3′ overhang. However, the end product is similar, as the top strand, after resection of the unmatched bases, and can simply be ligated to the top strand of the plasmid DNA without the need for further DNA synthesis. This type of repair, which occurs second, appears to be distinct from either traditional SDSA repair or ExACT, but bears similarities to SDSA. Another repair pathway example found in a different experiment that was created by dimerizing repair ssODNs is included in Supplemental Figure S2. It should be made clear, again, that this dimerization of the oligo is not thought to be the mechanism by which the ExACT pathway repairs DNA ends. The outcomes shown in these data are repair outcomes that are enriched in the population due to unforeseen problems with repair template design (i.e., the higher energy favorability of oligo dimerization compared to repair homology). However, these imprecise outcomes still indicate that both single-event gap bridging and multi-stage repair can occur, which further indicates that the ExACT model, as described previously, is incomplete in its description of DNA repair and must be updated to include these SDSA-like repair mechanisms.
References
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.-S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232.
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823.
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.-R.J.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229.
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239.
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826.
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J. RNA-programmed genome editing in human cells. eLife 2013, 2, e00471.
- Sternberg, S.H.; Redding, S.; Jinek, M.; Greene, E.C.; Doudna, J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014, 507, 62–67.
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015, 163, 759–771.
- Kim, H.; Kim, J.-S. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014, 15, 321–334.
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10502–10511.
- Brandsma, I.; Gent, D.C. Pathway choice in DNA double strand break repair: Observations of a balancing act. Genome Integr. 2012, 3, 9.
- Patsali, P.; Kleanthous, M.; Lederer, C.W. Disruptive Technology: CRISPR/Cas-Based Tools and Approaches. Mol. Diagn. Ther. 2019, 23, 187–200.
- Carvajal-Garcia, J.; Cho, J.E.; Carvajal-Garcia, P.; Feng, W.; Wood, R.D.; Sekelsky, J.; Gupta, G.P.; Roberts, S.A.; Ramsden, D.A. Mechanistic basis for microhomology identification and genome scarring by polymerase theta. Proc. Natl. Acad. Sci. USA 2020, 117, 8476–8485.
- Hanscom, T.; McVey, M. Regulation of Error-Prone DNA Double-Strand Break Repair and Its Impact on Genome Evolution. Cells 2020, 9, 1657.
- Truong, L.N.; Li, Y.; Shi, L.Z.; Hwang, P.Y.H.; He, J.; Wang, H.; Razavian, N.; Berns, M.W.; Wu, X. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7720–7725.
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257.
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355.
- Boroviak, K.; Doe, B.; Banerjee, R.; Yang, F.; Bradley, A. Chromosome engineering in zygotes with CRISPR/Cas9. Genesis 2016, 54, 78–85.
- Dow, L.E. Modeling Disease In Vivo With CRISPR/Cas9. Trends Mol. Med. 2015, 21, 609–621.
- Parekh-Olmedo, H.; Kmiec, E.B. Progress and prospects: Targeted gene alteration (TGA). Gene Ther. 2007, 14, 1675–1680.
- Engstrom, J.U.; Suzuki, T.; Kmiec, E.B. Regulation of targeted gene repair by intrinsic cellular processes. BioEssays 2009, 31, 159–168.
- Aarts, M.; te Riele, H. Progress and prospects: Oligonucleotide-directed gene modification in mouse embryonic stem cells: A route to therapeutic application. Gene Ther. 2011, 18, 213–219.
- Olsen, P.A.; Randol, M.; Krauss, S. Implications of cell cycle progression on functional sequence correction by short single-stranded DNA oligonucleotides. Gene Ther. 2005, 12, 546–551.
- Radecke, F.; Peter, I.; Radecke, S.; Gellhaus, K.; Schwarz, K.; Cathomen, T. Targeted chromosomal gene modification in human cells by single-stranded oligodeoxynucleotides in the presence of a DNA double-strand break. Mol. Ther. 2006, 14, 798–808.
- Engstrom, J.U.; Kmiec, E.B. DNA replication, cell cycle progression and the targeted gene repair reaction. Cell Cycle 2008, 1402–1414.
- Radecke, S.; Radecke, F.; Peter, I.; Schwarz, K. Physical incorporation of a single-stranded oligodeoxynucleotide during targeted repair of a human chromosomal locus. J. Gene Med. 2006, 8, 217–228.
- Andersen, M.S.; Sorensen, C.B.; Bolund, L.; Jensen, T.G. Mechanisms underlying targeted gene correction using chimeric RNA/DNA and single-stranded DNA oligonucleotides. J. Mol. Med. 2002, 80, 770–781.
- Rivera-Torres, N.; Strouse, B.; Bialk, P.; Niamat, R.A.; Kmiec, E.B. The Position of DNA Cleavage by TALENs and Cell Synchronization Influences the Frequency of Gene Editing Directed by Single-Stranded Oligonucleotides. PLoS ONE 2014, 9, e96483.
- Brachman, E.E.; Kmiec, E.B. Gene repair in mammalian cells is stimulated by the elongation of S phase and transient stalling of replication forks. DNA Repair 2005, 4, 445–457.
- Olsen, P.A.; Randol, M.; Luna, L.; Brown, T.; Krauss, S. Genomic sequence correction by single-stranded DNA oligonucleotides: Role of DNA synthesis and chemical modifications of the oligonucleotide ends. J. Gene Med. 2005, 7, 1534–1544.
- Ferrara, L.; Parekh-Olmedo, H.; Kmiec, E.B. Enhanced oligonucleotide-directed gene targeting in mammalian cells following treatment with DNA damaging agents. Exp. Cell Res. 2004, 300, 170–179.
- Bialk, P.; Sansbury, B.; Rivera-Torres, N.; Bloh, K.; Man, D.; Kmiec, E.B. Analyses of point mutation repair and allelic heterogeneity generated by CRISPR/Cas9 and single-stranded DNA oligonucleotides. Sci. Rep. 2016, 6, 32681.
- Davis, L.; Maizels, N. Homology-directed repair of DNA nicks via pathways distinct from canonical double-strand break repair. Proc. Natl. Acad. Sci. USA 2014, 111, E924–E932.
- Richardson, C.D.; Ray, G.J.; DeWitt, M.A.; Curie, G.L.; Corn, J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344.
- Paquet, D.; Kwart, D.; Chen, A.; Sproul, A.; Jacob, S.; Teo, S.; Olsen, K.M.; Gregg, A.; Noggle, S.; Tessier-Lavigne, M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 2016, 533, 125–129.
- Ling, X.; Xie, B.; Gao, X.; Chang, L.; Zheng, W.; Chen, H.; Huang, Y.; Tan, L.; Li, M.; Liu, T. Improving the efficiency of precise genome editing with site-specific Cas9-oligonucleotide conjugates. Sci. Adv. 2020, 6, eaaz0051.
- Renaud, J.B.; Boix, C.; Charpentier, M.; De Cian, A.; Cochennec, J.; Duvernois-Berthet, E.; Perrouault, L.; Tesson, L.; Edouard, J.; Thinard, R.; et al. Improved Genome Editing Efficiency and Flexibility Using Modified Oligonucleotides with TALEN and CRISPR-Cas9 Nucleases. Cell Rep. 2016, 14, 2263–2272.
- Hendel, A.; Bak, R.O.; Clark, J.T.; Kennedy, A.B.; Ryan, D.E.; Roy, S.; Steinfeld, I.; Lunstad, B.D.; Kaiser, R.J.; Wilkens, A.B.; et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015, 33, 985–989.
- Rivera-Torres, N.; Banas, K.; Bialk, P.; Bloh, K.M.; Kmiec, E.B. Insertional Mutagenesis by CRISPR/Cas9 Ribonucleoprotein Gene Editing in Cells Targeted for Point Mutation Repair Directed by Short Single-Stranded DNA Oligonucleotides. PLoS ONE 2017, 12, e0169350.
- Valerie, K.; Povirk, L.F. Regulation and mechanisms of mammalian double-strand break repair. Oncogene 2003, 22, 5792–5812.
- Davis, A.J.; Chen, D.J. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013, 2, 130–143.
- Parekh-Olmedo, H.; Ferrara, L.; Brachman, E.; Kmiec, E.B. Gene therapy progress and prospects: Targeted gene repair. Gene Ther. 2005, 12, 639–646.
- Radecke, F.; Radecke, S.; Schwarz, K. Unmodified oligodeoxynucleotides require single-strandedness to induce targeted repair of a chromosomal EGFP gene. J. Gene Med. 2004, 6, 1257–1271.
- Resnick, M.A. The repair of double-strand breaks in DNA: A model involving recombination. J. Theor. Biol. 1976, 59, 97–106.
- Nassif, N.; Penney, J.; Pal, S.; Engels, W.R.; Gloor, G.B. Efficient copying of nonhomologous sequences from ectopic sites via P-element-induced gap repair. Mol. Cell. Biol. 1994, 14, 1613–1625.
- Maher, R.L.; Branagan, A.M.; Morrical, S.W. Coordination of DNA replication and recombination activities in the maintenance of genome stability. J. Cell. Biochem. 2011, 112, 2672–2682.
- Holliday, R. The induction of mitotic recombination by mitomycin c in ustilago and saccharomyces. Genetics 1964, 50, 323–335.
- Allers, T.; Lichten, M. Differential timing and control of noncrossover and crossover recombination during meiosis. Cell 2001, 106, 47–57.
- DeWitt, M.A.; Magis, W.; Bray, N.L.; Wang, T.; Berman, J.R.; Urbinati, F.; Heo, S.-J.; Mitros, T.; Munoz, D.P.; Boffelli, D.; et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci. Transl. Med. 2016, 8, 360ra134.
- Park, S.H.; Lee, C.M.; Dever, D.P.; Davis, T.H.; Camarena, J.; Srifa, W.; Zhang, Y.; Paikari, A.; Chang, A.K.; Porteus, M.H.; et al. Highly efficient editing of the β-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res. 2019, 47, 7955–7972.
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211.
- Morrical, S.W. DNA-pairing and annealing processes in homologous recombination and homology-directed repair. Cold Spring Harb. Perspect. Biol. 2015, 7, a016444.
- Sansbury, B.M.; Wagner, A.M.; Tarcic, G.; Barth, S.; Nitzan, E.; Goldfus, R.; Vidne, M.; Kmiec, E.B. CRISPR-Directed Gene Editing Catalyzes Precise Gene Segment Replacement In Vitro Enabling a Novel Method for Multiplex Site-Directed Mutagenesis. Cris J. 2019, 2, 121–132.
- Sansbury, B.M.; Hewes, A.M.; Kmiec, E.B. Understanding the diversity of genetic outcomes from CRISPR-Cas generated homology-directed repair. Commun. Biol. 2019, 2, 458.
More