Cancer is a multifactorial disease with increasing incidence. There are more than 100 different cancer types, defined by location, cell of origin, and genomic alterations that influence oncogenesis and therapeutic response. This heterogeneity between tumors of different patients and also the heterogeneity within the same patient’s tumor pose an enormous challenge to cancer treatment.
- intra-tumor heterogeneity
- multi-omics technology
- cancer models
- patient-derived organoids
- personalized oncology
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
Despite all advancements in research and clinical practice, cancer remains a life-threatening disease with increasing incidence. Based on a prognosis by the WHO in 2018, cancer incidence is expected to double to about 37 million new cancer cases by 2040 [1].
Effective disease management is critical to cancer treatment. Current cancer treatments have made enormous progress from the first cytotoxic agents aiming at replicating cells via targeted therapies selectively aiming at genomic aberrant pathways like cetuximab for the treatment of advanced colorectal cancer to immuno-targeted drugs like ipilimumab for the treatment of malignant melanoma [2,3][2][3].
Despite the progress in medical oncology, most cancers are still treated by surgical resection of the tumors if accessible [4]. Pivotal to subsequent adjuvant or neo-adjuvant chemotherapy is the application of strict guideline protocols (S3 guidelines in Europe). These protocols are based on studies of large patient cohorts with similar cancers. This approach has led to a significant increase in progression-free (PFS) and overall survival (OAS) of patients, yet a majority of patients does not fully benefit from the administered treatment regimen [5]. The reason for therapeutic regimens falling short in those patients is the fact that every cancer comes with an individual, virtually unique genetic landscape [6].
Until it will become possible to faithfully predict the individual outcome of a specific treatment, oncologists and patients alike experience painful uncertainty regarding therapy success at the start and course of treatment. Even worse, both parties know that even when the tumor does not regress, the dire side effects of treatment will still impact the patient’s quality of life.
It is the aim of precision oncology to overcome this dilemma. Understanding the interplay between the unique characteristics of a patient’s tumor and the medical treatment and how it can be tailored to the individual properties of the tumor is a major focus in translational cancer research. Today, cancer precision medicine mostly aims at matching specific tumor mutations with drugs targeting aberrant oncogenic pathways to provide individualized treatment options relying on small organic compounds and/or monoclonal antibodies [7]. While most cancers harbor multiple oncogenic mutations, preclinical and clinical data support the idea that many cancers are sensitive to inhibition of single oncogenes, a concept referred to as ‘oncogene addiction’ [8].
This mutation-driven approach to cancer precision medicine is also applied to pre-therapeutic cohort stratification, which has subsequently led to the concept of conditional approvals, i.e., certain, targeted cancer therapeutics are only approved for patients with a defined set of specific mutations.
The precedent of a targeted cancer therapeutic with conditional approval is the anti-epidermal growth factor receptor (EGFR) antibody cetuximab (Erbitux®). In colorectal cancer (CRC), examination of molecular alterations indicated that mutations in KRAS, which is downstream from EGFR in the RAS-MEK-ERK signaling pathway, interfered with this therapy [9,10][9][10]. Cetuximab is therefore only relevant for RAS wildtype tumors. Following the success of cetuximab, other small molecules, biosimilar and monoclonal antibodies were investigated and successfully applied in the clinic. Today, our arsenal of targeted drugs in oncology comprises 84 approved agents [11].
Yet, cancer is far from being conquered, reflected by the fact that cancer is the second leading cause of death worldwide accounting for an estimated 9.6 million deaths in 2018 [12]. In addition, it becomes more and more evident that the genetic approach to cancer precision medicine alone is not sufficient to predict individual treatment response. This is mainly due to intra-tumor heterogeneity, which is currently not sufficiently taken into consideration.
2. Intra-Tumor Heterogeneity—The Challenge of Treating “Many Cancers in One”
The biology of cancer is complex and not yet fully understood. During malignant transformation tumors may acquire increasingly aggressive features and over time increase their metastatic potential and propensity to gain treatment resistance [13][14]. These hallmark features develop by clonal evolution which is fueled by the complex interplay of cancer cells and their microenvironment [15]. This unique composition of any given tumor is one of the biggest clinical challenges in modern oncology [16][17].
At the time of diagnosis, the tumor usually has passed millions to billions of aberrant cell divisions that frequently lead to genetic instability and genomic and epigenetic heterogeneity [18]. The path of malignant transformation from benign hyperplasia (non-cancerous) to malignant (cancerous) may follow different routes of clonal evolution [19]. The later the tumor has been diagnosed the higher degree of intra-tumoral diversity is expected [13]. Heterogeneity happens latitudinally between tumors from different patients (inter-patient heterogeneity) and longitudinally in the tumor (and metastases) of the same patient (intra-patient heterogeneity) [20]. A well-described example for heterogeneity is the hepato-cellular carcinoma (HCC) with a high degree of longitudinal heterogeneity between patients as well as latitudinal heterogeneity within the same tumor of an individual patient [21].
An emerging field is the immunogenic heterogeneity, which is common in liver tumors but not limited to this tumor entity [22][23]. Tumors from different patients show a different degree of immune cell infiltration and immune cell composition [23]. Immunologically “hot” tumors present high levels of T cell infiltration [24]. In consequence, these tumors are more susceptible to immune-checkpoint inhibitor treatment as compared to immunologically “cold” tumors [24]. This immunogenic heterogeneity impacts treatment outcome. There are multiple studies ongoing which aim to define a consensus classification for molecular (immune) subclasses [22][25].
Intra-patient heterogeneity or intra-tumor heterogeneity (ITH) has been proven for solid tumors and usually refers to genetic changes within cell subpopulations that form the tumor mass after multiple cell divisions and proliferation during tumor growth [26][27][28][29]. Thus, solid tumors can be described as heterogeneous neoplasms comprised of different types of cells. A representative solid tumor is composed of malignant cells, communized with mesenchymal cells, endovascular cells, and immune cells creating the tumor microenvironment [30].
ITH is the result of rather complex events and context, related to different causes and different outcome patterns. There are different types of ITH: morpho-histological [31][32], clonal [15] and nonclonal ITH [33][34][35].
Morpho-histological ITH is represented by different morphological structures, is related to different levels of differentiation and/or correlates with different molecular alterations, which was shown in lung adenocarcinomas where the mutant allele frequencies were higher in solid areas of the same tumor [36]. In breast cancer for example, morphological ITH was found to be associated with epithelial–mesenchymal transition (EMT) and stemness of cancer cells [32].
Nonclonal ITH derives from microenvironment interaction which makes the clinical approach challenging [31]. The tumor microenvironment is in constant chemical and physical interaction with the actual tumor. These interactions vary between the different areas of the primary tumor. Further, these interactions may not only fuel heterogeneity amongst the tumor cells, but also the stroma can become increasingly heterogeneous [33]. Besides paracrine signaling of cancer and stroma cells, the interaction with the different types of collagen within the stroma may impact therapy success [34][35].
Clonal ITH derives from genomic instability [37]. Different individual tumors of the same entity may undergo very different paths of clonal evolution, which was shown in liver cancer [38]. ITH is the basis for the selection of the fittest clones, a key step in clonal evolution [15].
Following the development of new analytic technologies, recent data support the clonal evolution model as the main theoretical basis of tumor heterogeneity [19][39].
A recent study from Yang et al. in patients with glioblastoma (GBM) provides a good understanding of intra-tumor heterogeneity and its impact on disease progression and recurrence. Regions from within the tumor were sequenced as well as blood-derived circulating tumor DNA from patients with primary and recurrent GBM [40]. They observed high intra-tumor heterogeneity at the levels of both somatic gene mutations and chromosomal copy numbers. In total, they analyzed over 1000 genes that are involved in tumorigenesis and cancer progression [40].
Another example revealing substantial ITH in a solid tumor is a recent study by Schumacher et al. using multi-region sampling of colorectal cancer. They generated five patient-derived three-dimensional (PD3D) “sibling” in vitro cultures from a single colorectal tumor and modelled consequences of ITH on tumor cell growth and drug response [41]. The heterogeneity of tumor tissues and PD3D sibling cultures was further evaluated at DNA and mRNA levels. Using cancer panel-sequencing, common mutations were detected (KRAS
, PIK3CAH
, and TP53
) in the tumor tissue-of-origin and the separate sibling cultures [41]. They found an additional homozygous SMAD4
mutation in two out of the five regions which might be responsible for different drug responses of the different cultures. Not only did drug responses of sibling cultures show up to a 30-fold difference, but also substantial heterogeneity in mRNA expression of target genes of ERK/MAPK, PI3K and mTOR signaling pathways was observed [41]. Such investigations are crucial for the acceptance of testing patient-derived models as they aim to understand the heterogeneity of tumors in the clinical context. This will offer easy access to molecular “toolboxes” in translational oncology to learn how to overcome treatment resistance.
The impact of ITH on drug resistance and targeted therapy strategies has been demonstrated using multiple-site profiling in solid tumors and is regarded as a paradigm shift in cancer care [42]. In contrast, only a limited number of studies addressed ITH in non-solid tumors [43][44][45][46]. Wogsland et al. studied intra- and inter-tumor heterogeneity of follicular lymphoma (FL), a B-cell malignancy, by using mass cytometry to obtain deep profiling of cell subsets [43]. This study allowed the identification of biologically relevant features including tumor heterogeneity and loss of non-malignant B-cell subsets. Additional proteins with a high variability among lymphoma cells have been identified in the same tumor [43].
In a proof-of-concept study, Araf et al. performed a combination of whole-exome and targeted deep sequencing of spatially and temporally separated biopsies from patients with follicular lymphoma [44]. Their results suggest that the analyzed tumors consisted of multiple subclones. Different tumor subpopulations dynamically circulated in the plasma with increasing heterogeneity during malignant transformation. Yet, their data did not show an association between increasing genetic ITH and therapy success [44]. Nevertheless, these results warrant further studies about ITH in liquid tumors to understand its relevance in the clinic.
In summary, ITH has been identified as being causal for the often-observed failure of current tumor therapies, especially in solid tumors. In addition, ITH might limit the reproducibility in clinical cancer research and precise diagnostic evaluation of tumors [47]. Therefore, ITH is a major challenge of cancer treatment which needs to be considered in precision cancer medicine.
3. Integration of Intra-Tumor Heterogeneity into Multi-Layered Personalized Cancer Therapy
Regardless of the tumor evolution that is causing ITH, the existence of multiple distinctive cell populations in the same tumor has strong clinical implications [48]. The diagnosis of primary and metastatic tumors is typically based on a single biopsy representing only a snapshot of ongoing tumor evolution and may be compromised by ITH [49]. The studies described above show that a sole biopsy does not accurately capture the tumor’s genetic and phenotypic heterogeneity [28][46]. Therefore, multimodal strategies have to be taken into account to ensure patients receive effective and targeted therapy. In addition, it is necessary to develop new tools to study heterogeneity, and to identify new biomarkers of tumor heterogeneity. Further, besides focusing on clonal heterogeneity, nonclonal phenotypic heterogeneity should be taken into consideration, i.e., the fact that some cells respond to broad, environmental perturbations and drug treatments by conversion to many other cell states, including stem-like, resistant cell phenotypes [50]. Finally, suitable models are needed that also take the effect of the tumor microenvironment into account.
3.1. Current Approaches to Analyze ITH from Tumor Samples
Still, most efforts of molecular tumor boards predominantly focus on genomic sequence information to diagnose patients, to predict the individual risk of developing cancer, and to assess whether specific treatments are suitable (i.e., likely to be successful) [7]. The concept of ITH is only rarely covered in these boards.
Major advances in the concept of tumor heterogeneity have come with the implementation of high-throughput genomic sequencing technologies. These allow the profiling of chromosomal and point mutations of neoplasms, with genomic data being most commonly used for molecular cancer diagnostics for rational clinical decision making [32].
However, intra-tumoral heterogeneity results from much more complex interplay on different levels. Genetic, epigenetic, and protein modifications are determining the phenotype of a given tumor. Plasticity allows for adaption in response to environmental factors by modulation of cellular properties [51]. These complexities are very well put into conceptual context by Brock and Huang [52]. Recent studies showed that only about 10% of cancer patients benefit from genomic sequencing of their tumor [53]. This argues the use of a single technology for decision making but suggests a multi-omics approach to provide deeper integration of precision oncology into the clinic in order to pave the way for tailored therapy schedules for the benefit of patients and doctors alike.
3.1.1. Genomic Approaches to ITH and Precision Oncology
Today, precision oncology is based on the assumption that tumor treatment is more effective when selecting a target-specific therapy that matches the genetic or epigenetic changes observed in a single tumor of a cancer patient [13].
Since the genomic revolution more than 30 years ago, array-based and sequencing approaches have been used extensively to identify and characterize the molecular events underlying cancer development and progression [54]. Today, many of these learnings are widely used for diagnostics. Large multinational consortia like “The Cancer Genome Atlas” (TCGA) initiative provide datasets of individual tumors and tissues from thousands of patients from a wide array of cancers providing insights into the complexity of cancer [55]. It was found that mutations within the same tumor differ enormously in their allelic frequencies [56]. Based on the grouping of allelic frequencies, an estimate of the genetically different “clones” of cancer cells can be made for each tumor [56].
The presence of multiple clones within the same tumor sparked an ongoing discussion about the need of multi-regional sampling to discover ITH in tumors. Surprisingly, Zhang et al. concluded that complete assessment of ITH complexity may not require sampling in multiple regions [57]. They showed for lung adenocarcinoma that a single biopsy may be sufficient to capture the majority of mutations if ultra-deep sequencing is performed [57]. This is in stark contrast to other studies like the one by Gerlinger et al. [58]. Multiregional sequencing on samples obtained from primary renal carcinomas and associated metastatic sites demonstrated substantial ITH, with several mutations in certain cancer genes being restricted to separated tumor domains [58]. These studies suggest that a single biopsy may not be suitable for the identification of all cancer gene mutations of a tumor, thus providing an incomplete view of potential targets for cancer therapy [13][58][59].
Surprisingly, several clinical trials driven by genomics have been resulting in positive response rates of only 5–10% [53]. Among those studies, the most successful study was the WINTHER trial (NCT01856296). During the six-year study, patients were stratified to different treatments based on DNA-sequencing from fresh biopsy-derived tissue (arm A; 236 gene panel) vs. RNA expression analysis (arm B; comparing tumor to normal). Based on the sequencing results, treatment recommendations were made by a multi-national “clinical management committee” with subsequent monitoring of the applied therapy by oncologists. Treatment scores were calculated for each patient based on an array of clinical parameters. Within the patient cohort included in the study, stable disease (≥6 months) and partial or complete response was 26.2% (arm A: 23.2%; arm B: 31.6%) compared to previous treatment regimen progression-free survival of 22.4% [60]. The most important predictor of successful treatment was actually fewer previous therapies, while genomic and transcriptomic profiling could only marginally improve treatment recommendations and patient outcome [61].
While genomic analysis is invaluable for our current understanding of cancer and the development of better therapies, sequencing alone is not sufficient for tailored therapies. These molecular snapshots cannot answer clinically relevant questions: does a specific alteration occur in all cells or just in a small part of the tumor? How many regions of a tumor should be sequenced to get a representative result? In addition, the presence or absence of a specific mutation is not always sufficient to predict the effect of a targeted therapy. This is well documented in colorectal cancer, where
mutations are used to estimate the potential success of anti-EGFR treatment [62]. While anti-EGFR competitors like cetuximab or panitumumab correspond with a longer overall survival of patients with
wild-type, not all
wild-type patients benefit from upstream RAS inhibition due to the tissue context [63][64][65][66][67][68]. Patients with right hemi colic cancers do not benefit from anti-EGFR therapy irrespective of RAS status [69]. Current guidelines take such spatial heterogeneity into account, and therefore suggest the use of cetuximab or panitumumab only for patients with
3.1.2. Proteomic Approaches
During the last few decades, not only have sequencing technologies advanced significantly, but other technologies such as proteome and metabolome analyses have also become more suitable for a deeper characterization of tumor cells and their functional abnormalities [72][73].
Transcriptomics capture the quantity of the immediate product of a cell’s genome at a given timepoint. It is a snapshot of the genomic expression at a certain time, or the change thereof over time or under different conditions [74]. In contrast, proteomics dissect the cellular protein composition, the result of gene translation [75]. Initial large-scale studies of cellular proteomes showed a relatively low correlation between protein expression levels and corresponding mRNA expression levels [64].
As on the genomic level, protein function is mediated and altered by multiple mechanisms. Posttranslational modifications of proteins (e.g., phosphorylation) are often required for their activity or signaling. Folding and posttranslational processing of proteins is a prerequisite for the formation of multi-protein complexes, which in turn are necessary to act as molecular machines, performing almost all cellular functions. To add another layer of complexity, the cellular location of a specific protein often determines whether the protein is active or inactive, or if a protein complex is successfully formed. In cancer proteomics, the TCGA Consortium represents the first large-scale effort to profile the tumor proteome [76]. However, the analysis was performed using reverse phase protein arrays, and was therefore limited to the targeted analysis of a few hundred proteins.
Despite the complexity, proteomic approaches have led to the identification of specific biomarkers in ovarian cancer [65] and identification of molecular subgroups in breast cancer [66]. Proteomic data used in precision oncology has helped to correctly predict drug sensitivity and resistance [67][68]. Cancer proteomics can therefore be seen as complementing the traditional immunohistochemical classification of tumor types, such as the characterization of estrogen receptor expression in breast cancers [77][78].
3.1.3. Metabolomic Approaches
Metabolites are the products of cellular processes, which in turn are driven by proteins, mostly enzymes. Therefore, changes in metabolites mirror changes in the activity of enzymes and proteins and may pose as ideal biomarkers [79]. Due to their accessibility, many metabolomic analyses are performed on plasma or serum samples from patients used in diagnosis without requiring an invasive intervention to obtain tumor tissue [80][81]. Yet, this analysis of individual levels of metabolites makes it difficult to determine universal levels and individual changes for a particular tumor entity. Currently, only few studies have been conducted, and validation is pending [82]. Among the few examples of a clinically relevant metabolomic approach is the use of metabolites for the identification of altered carbohydrates in acute myeloid leukemia [83], as well as unsaturated free fatty acids in colorectal cancer [84].
Given the limitation of current knowledge on metabolomics in cancer in general, we are still far away from understanding how it may contribute to a better understanding of ITH from measuring metabolomic changes in peripheral blood and how this is representative for the complexity of the tumor tissue.
Nevertheless, with the rise of genomic medicine and in-depth characterization of the individual tumor mutation landscape, a better understanding of tumor complexity and ITH suggest that the ‘precision medicine’ paradigm of cancer therapy requires multi-modal treatment to be personalized to the individual patient.
3.2. Suitable In Vitro Strategies for Modeling Intra-Tumor Heterogeneity
Utilizing cancer models may complement or even supplement the approaches described above. ITH, the cellular interactions and the tumor’s molecular alterations could be investigated in cancer models derived from patients’ samples to study the underlying mechanisms and/or learn how ITH may direct clinical decision making.
Cancer models are naturally existing or artificially induced systems that share characteristics with human cancers [85]. Experimental systems for the study of human cancer include genetically engineered mouse models (GEMMs) [86][87][88][89], two-dimensional (2D) cell lines [90], patient-derived organoids (PDO) [91][92][93][94][95], and patient-derived xenografts (PDX) [95][96] to study biochemical or genetic pathways and pathology of cancer. These in vivo and in vitro cancer models have been invaluable for our current understanding of cancer development and progression, as well as for therapy development. Further, these models are moving into focus regarding their potential use in cancer precision and/or personalized medicine [97][98].
Given the complexity and heterogeneity of cancer, a crucial question to be asked is whether these models are feasible to capture and investigate ITH.
GEMMs are created by inducing specific mutations in oncogenes and/or tumor suppressor genes and can be used to monitor tumorigenesis in vivo, but are limited by species differences in oncogenic pathogenesis, the shorter lifespan of mice, and often by the artificial simultaneous introduction of several oncogenic driver events [88][89].
Traditional 2D cell lines grow as monolayer, cultured on flat and rigid substrates [91]. They have the advantage that they have once been derived from a cancer patient and are easier to manipulate in the laboratory, but they cannot completely replicate the environment of the patient tumor. Even within the same cancer model, data between laboratories are often irreproducible [99][100]. Nevertheless, cancer 2D cell lines have been used not only for in vitro but also for in vivo experiments, for example to generate xenograft models by subcutaneous injection of cancer cell lines into immunodeficient mice [101].
Patient-derived xenografts (PDX) are generated by implantation of cancerous tissue from a patient’s tumor either under the skin (ectopic) or into the organ of tumor origin (orthotopic) and are most commonly used for preclinical drug development [102][103][104].
Physiologically, cells grow in three dimensions (3D) to form discrete tissue and organ structures [105][106]. PDO models, growing in 3D, have been shown to reliably recapitulate the architecture of the donor tissue and to preserve its genomic background, therefore providing a highly relevant physiological system [107]. With their optimal conditions for cellular proliferation, differentiation, and responsiveness to chemo- and targeted therapeutics, they recapitulate the functional tumor phenotype, including its ITH [41].
The impact of adding a third culture dimension on the cellular drug response has been shown by Koch et al. [108]. They compared the response of 2D colorectal cancer cell lines and 3D CRC cell line-derived spheroids to irradiation and chemotherapy [108]. 3D CRC cell line cultures were more resistant to irradiation and chemotherapy, such as 5-FU and cisplatin, than their 2D counterparts [108]. This must be taken into account when translating the results into clinical setting. Therefore, PDOs are increasingly used for studying tumor biology and the effects of targeted therapies [109].
Schütte et al. investigated a colorectal cancer biobank comprising PDX models and PDO models, which were treated with clinically relevant compounds [95]. They showed that PDOs recapitulate many of the genetic and transcriptomic features of the donor tumors whereas clonal discordances found at early passages were attributed to ITH [95]. Ben-David et al. showed that the genomic and transcriptomic heterogeneity of cell lines impairs our ability to evaluate new therapeutics [110]. Their results support efforts to systematically develop PDO models to reduce the reliance on poorly defined cell lines that were established before the next generation sequencing era [110]. Further, Vlachogiannis et al. showed that PDOs can recapitulate patient responses in the clinic and could therefore be implemented as models for personalized oncology [111].
An additional advantage of PDO cultures is that they can be used in high-throughput drug screens. Such screens can be composed of multiple samples from the same tumor, thereby taking ITH into account [41][94].
compares this approach with a PDX approach aiming to test the same number of tumor samples and compounds to illustrate the time- and cost-effectiveness of the organoid system.
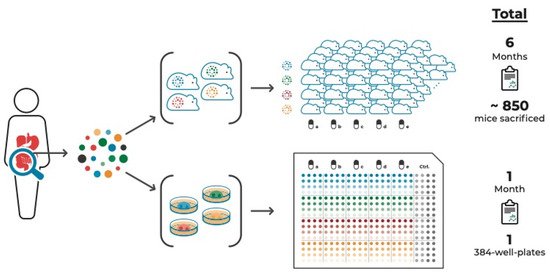
PDO vs. PDX cancer models to study a tumor’s drug response incorporating intra-tumor heterogeneity. Multi-regional sampling of a tumor is required to take ITH into account when assessing the tumor’s drug response. Compared to multi-regional PDX models, multi-regional PDO-based pre-therapeutic drug screenings are significantly more time- and cost-effective. These PDO-based screens, in addition to amplicon sequencing of multiple areas of tumor tissue and derived cell culture models, combined with targeted proteomic approaches, are both feasible and available within a timeframe that allows discussing guided treatment options.
Nevertheless, PDO are an in vitro culture system, therefore lacking, for example, liver or kidney clearance or liver pro-drug activation mechanism. Further, only the effect of the drug on the tumor (i.e., tumor-derived organoid) itself can be assessed, omitting potential toxicity on other organs or indirect effects asserted on, e.g., vasculature or hormone production in the pituitary gland. These aspects need to be taken into consideration for experimental design.
It is known that the microenvironment with tumor-surrounding and infiltrating cells, including fibroblasts and immune cells, have a major impact on drug response [112][113]. Another significant advantage of the organoid model system is the ability to study the interaction of cancer organoids with other specific cell types that can be introduced into a direct or indirect co-culture system. Indirect co-culture systems are based on the use of cell conditioned media. They are simple to apply and are therefore often used for in vitro experiments [114]. However, they are not suitable for investigating the effects of cell contacts between cancer cells and stroma [115]. Direct co-culture models are a closer representation of the in vivo scenario. Since the tumor microenvironment plays a critical role in tumorigenesis, 3D co-culture systems are used, including not only cancer cells but also stromal cells [115]. Cancer cells were localized in a defined area within a stromal cell matrix to study the cytotoxic effect of anticancer drugs on both tumor and normal cells in the same system [116].
Since antibodies against immune checkpoint proteins/receptors have shown clear clinical benefit for patients with advanced cancer, including melanoma, non-small cell lung cancer (NSCLC), and mismatch repair deficient (dMMR) colorectal cancer, organoid co-culture systems including immune cells are moving into the spotlight of current in vitro application [117][118][119][120][121][122][123][124][125][126]. Dijkstra et al. established a co-culture system of autologous tumor organoids and peripheral blood lymphocytes of patients to induce and analyze tumor-specific T cell responses for mismatch repair deficient colorectal cancer and non-small cell lung cancer in a personalized manner [118]. Klein et al. demonstrated the advantages of co-culture systems of GBM organoids and human immune cells, to investigate not only immune–tumor interactions, but also to explore current and novel immunotherapies, such as adoptive T cell transfer, immune checkpoint inhibitors, or oncolytic viruses [127].
Another promising technology for studying ITH is Organ-on-a-Chip (OoC), a culture model to mimic complex and dynamic in vivo microenvironments [128]. An OoC is a multi-channel 3D microfluidic biochip, which recapitulates the activity, mechanism, and pathophysiological reaction of single-organ and multi-organ systems [129]. It is a useful tool in controlling spatial arrangement of cell growth and fluids within micrometer-sized channels, which may be used to increase the physiological relevance of tumor models [130]. OoC technology is expected to offer effective solutions to investigate the effects of drugs, as well as the causes of diseases and personalized therapeutic treatments [131][132][133].
The development of a multi-organoid platform that consists of patient-specific tumor organoids is currently in process. It is intended to offer the opportunity to test the efficiency of drug therapies designed based on genetic profiling. Skardal et al. generated a circulatory system with multiple tissue organoid sites by using microfluidic chip devices and used them to visualize and track tumor progression and kinetics of metastasis formation to distant site in vitro [132][133]. In combination with the even more complex body-on-a-chip platform, these personalized on-a-chip systems will be improved even further [132][133].
This very promising technology offers great potential for in vivo tumor-like model systems to enable personalized drug screenings before treating patients and monitoring of organ systems in the OoC device for side effects at the same time. This new technology is still in development but has the potential to improve cancer treatment outcomes and patient care dramatically.
In summary, there are various suitable preclinical cancer models each with its own limitations. Despite this, these models are an attractive alternative or addition and have the potential to augment genetic and multi-omics approaches when considering ITH for precision cancer medicine.
References
- WHO. Report on Cancer: Setting Priorities, Investing Wisely and Providing Care for All; World Health Organization: Geneve, Switzerland, 2020.
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723.
- Shankaran, V.; Obel, J.; Benson, A.B. Predicting response to EGFR inhibitors in metastatic colorectal cancer: Current practice and future directions. Oncologist 2010, 15, 157–167.
- Tohme, S.; Simmons, R.L.; Tsung, A. Surgery for cancer: A trigger for metastases. Cancer Res. 2017, 77, 1548–1552.
- Roser, M.; Ritchie, H. Cancer. Available online: (accessed on 14 October 2020).
- Pich, O.; Muiños, F.; Lolkema, M.P.; Steeghs, N.; Gonzalez-Perez, A.; Lopez-Bigas, N. The mutational footprints of cancer therapies. Nat. Genet. 2019, 51, 1732–1740.
- Krzyszczyk, P.; Acevedo, A.; Davidoff, E.J.; Timmins, L.M.; Marrero-Berrios, I.; Patel, M.; White, C.; Lowe, C.; Sherba, J.J.; Hartmanshenn, C.; et al. The growing role of precision and personalized medicine for cancer treatment. Technology 2019, 6, 79–100.
- Weinstein, I.B.; Joe, A.; Felsher, D. Oncogene addiction. Cancer Res. 2008, 68, 3077–3080.
- Misale, S.; Nicolantonio, F.D.; Sartore-Bianchi, A.; Siena, S.; Bardelli, A. Resistance to anti-EGFR therapy in colorectal cancer: From heterogeneity to convergent evolution. Cancer Discov. 2014, 4, 1269–1280.
- Emburgh, B.O.V.; Sartore-Bianchi, A.; Nicolantonio, F.D.; Siena, S.; Bardelli, A. Acquired resistance to EGFR-targeted therapies in colorectal cancer. Mol. Oncol. 2014, 8, 1084–1094.
- Abramson, R. Overview of Targeted Therapies for Cancer. My Cancer Genome. Available online: (accessed on 30 January 2021).
- Wild, C.P.; Weiderpass, E.; Stewart, B.W. World Cancer Report: Cancer Research for Cancer Prevention; IARC Publications: Lyon, France, 2020; ISBN 978-92-832-0448-0.
- Caravagna, G.; Giarratano, Y.; Ramazzotti, D.; Tomlinson, I.; Graham, T.A.; Sanguinetti, G.; Sottoriva, A. Detecting repeated cancer evolution from multi-region tumor sequencing data. Nat. Methods 2018, 15, 707–714.
- McGranahan, N.; Swanton, C. Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell 2017, 168, 613–628.
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306.
- Fisher, R.; Pusztai, L.; Swanton, C. Cancer heterogeneity: Implications for targeted therapeutics. Brit. J. Cancer 2013, 108, 479–485.
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta BBA Rev. Cancer 2010, 1805, 105–117.
- Schmitt, M.W.; Loeb, L.A.; Salk, J.J. The influence of subclonal resistance mutations on targeted cancer therapy. Nat. Rev. Clin. Oncol. 2016, 13, 335–347.
- Ding, L.; Raphael, B.J.; Chen, F.; Wendl, M.C. Advances for studying clonal evolution in cancer. Cancer Lett. 2013, 340, 212–219.
- Garattini, S.; Nerini, I.F.; D’Incalci, M. Not only tumor but also therapy heterogeneity. Annals of Oncology 2017, 29, 13–19.
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018.
- Kurebayashi, Y.; Ojima, H.; Tsujikawa, H.; Kubota, N.; Maehara, J.; Abe, Y.; Kitago, M.; Shinoda, M.; Kitagawa, Y.; Sakamoto, M. Landscape of immune microenvironment in hepatocellular carcinoma and its additional impact on histological and molecular classification. Hepatology 2018, 68, 1025–1041.
- Zhang, S.-C.; Hu, Z.-Q.; Long, J.-H.; Zhu, G.-M.; Wang, Y.; Jia, Y.; Zhou, J.; Ouyang, Y.; Zeng, Z. Clinical implications of tumor-infiltrating immune cells in breast cancer. J. Cancer 2019, 10, 6175–6184.
- Vareki, S.M. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J. Immunother. Cancer 2018, 6, 157.
- Zhang, Q.; Lou, Y.; Yang, J.; Wang, J.; Feng, J.; Zhao, Y.; Wang, L.; Huang, X.; Fu, Q.; Ye, M.; et al. Integrated multiomic analysis reveals comprehensive tumour heterogeneity and novel immunophenotypic classification in hepatocellular carcinomas. Gut 2019, 68, 2019.
- Sowalsky, A.G.; Ye, H.; Bhasin, M.; Allen, E.M.V.; Loda, M.; Lis, R.T.; Montaser-Kouhsari, L.; Calagua, C.; Ma, F.; Russo, J.W.; et al. Neoadjuvant-Intensive Androgen Deprivation Therapy Selects for Prostate Tumor Foci with Diverse Subclonal Oncogenic Alterations. Cancer Res. 2018, 78, 4716–4730.
- Nowell, P. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28.
- Kwon, S.M.; Budhu, A.; Woo, H.G.; Chaisaingmongkol, J.; Dang, H.; Forgues, M.; Harris, C.C.; Zhang, G.; Auslander, N.; Ruppin, E.; et al. Functional genomic complexity defines intratumor heterogeneity and tumor aggressiveness in liver cancer. Sci. Rep. 2019, 9, 16930.
- Sun, X.; Yu, Q. Intra-tumor heterogeneity of cancer cells and its implications for cancer treatment. Acta Pharmacol. Sin. 2015, 36, 1219–1227.
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773.
- Stanta, G.; Bonin, S. A practical approach to tumor heterogeneity in clinical research and diagnostics. Pathobiology 2018, 85, 7–17.
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41.
- Strickaert, A.; Saiselet, M.; Dom, G.; Deken, X.D.; Dumont, J.E.; Feron, O.; Sonveaux, P.; Maenhaut, C. Cancer heterogeneity is not compatible with one unique cancer cell metabolic map. Oncogene 2017, 36, 2637–2642.
- Erkan, M.; Erkan, M.; Hausmann, S.; Michalski, C.W.; Fingerle, A.A.; Dobritz, M.; Kleeff, J.; Friess, H. The role of stroma in pancreatic cancer: Diagnostic and therapeutic implications. Nat. Rev. Gastroentero 2012, 9, 454.
- Valkenburg, K.C.; Groot, A.E.D.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381.
- Stanta, G.; Bonin, S. Overview on clinical relevance of intra-tumor heterogeneity. Front. Med. 2018, 5, 85.
- Loeb, L.A. Human cancers express mutator phenotypes: Origin, consequences and targeting. Nat. Rev. Cancer 2011, 11, 450–457.
- Losic, B.; Craig, A.J.; Villacorta-Martin, C.; Martins-Filho, S.N.; Akers, N.; Chen, X.; Ahsen, M.E.; Von Felden, J.; Labgaa, I.; DʹAvola, D.; et al. Intratumoral heterogeneity and clonal evolution in liver cancer. Nat. Commun. 2020, 11, 291.
- Swanton, C. Intratumor heterogeneity: Evolution through space and time. Cancer Res. 2012, 72, 4875–4882.
- Yang, C.; Tan, Y.; Li, S.; Zhou, J.; Wang, Q.; Wang, Y.; Xie, Y.; Chen, L.; Li, J.; Fang, C.; et al. Genomic landscapes by multiregion sequencing combined with circulation tumor DNA detection contribute to molecular diagnosis in glioblastomas. Aging 2019, 11, 11224–11243.
- Schumacher, D.; Andrieux, G.; Boehnke, K.; Keil, M.; Silvestri, A.; Silvestrov, M.; Keilholz, U.; Haybaeck, J.; Erdmann, G.; Sachse, C.; et al. Heterogeneous pathway activation and drug response modelled in colorectal-tumor-derived 3D cultures. PLoS Genet. 2019, 15, e1008076.
- Lim, Z.-F.; Ma, P.C. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J. Hematol. Oncol. 2019, 12, 134.
- Wogsland, C.E.; Greenplate, A.R.; Kolstad, A.; Myklebust, J.H.; Irish, J.M.; Huse, K. Mass cytometry of follicular lymphoma tumors reveals intrinsic heterogeneity in proteins including HLA-DR and a deficit in nonmalignant plasmablast and germinal center B-cell populations. Cytom. Part B Clin. Cytom. 2017, 92, 79–87.
- Araf, S.; Wang, J.; Korfi, K.; Pangault, C.; Kotsiou, E.; Rio-Machin, A.; Rahim, T.; Heward, J.; Clear, A.; Iqbal, S.; et al. Correction: Genomic profiling reveals spatial intra-tumor heterogeneity in follicular lymphoma. Leukemia 2019, 33, 1540.
- Gawad, C.; Koh, W.; Quake, S.R. Dissecting the clonal origins of childhood acute lymphoblastic leukemia by single-cell genomics. P. Natl. Acad. Sci. USA 2014, 111, 17947–17952.
- Farmanbar, A.; Firouzi, S.; Makałowski, W.; Kneller, R.; Iwanaga, M.; Utsunomiya, A.; Nakai, K.; Watanabe, T. Mutational intratumor heterogeneity is a complex and early event in the development of adult T-cell leukemia/lymphoma. Neoplasia 2018, 20, 883–893.
- Stanta, G.; Jahn, S.W.; Bonin, S.; Hoefler, G. Tumour heterogeneity: Principles and practical consequences. Virchows Arch. 2016, 469, 371–384.
- Gambara, G.; Gaebler, M.; Keilholz, U.; Regenbrecht, C.R.A.; Silvestri, A. From chemotherapy to combined targeted therapeutics: In vitro and in vivo models to decipher intra-tumor heterogeneity. Front. Pharmacol. 2018, 9, 77.
- Brouwer, A.; Laere, B.D.; Peeters, D.; Peeters, M.; Salgado, R.; Dirix, L.; Laere, S.V. Evaluation and consequences of heterogeneity in the circulating tumor cell compartment. Oncotarget 2015, 7, 48625–48643.
- Inde, Z.; Dixon, S.J. The impact of non-genetic heterogeneity on cancer cell death. Crit. Rev. Biochem. Mol. 2017, 53, 1–16.
- Hinohara, K.; Polyak, K. Intratumoral heterogeneity: More than just mutations. Trends Cell Biol. 2019, 29, 569–579.
- Brock, A.; Huang, S. Precision oncology: Between vaguely right and precisely wrong. Cancer Res. 2017, 77, 6473–6479.
- Malone, E.R.; Oliva, M.; Sabatini, P.J.B.; Stockley, T.L.; Siu, L.L. Molecular profiling for precision cancer therapies. Genome Med. 2020, 12, 8.
- MacConaill, L.E. Existing and emerging technologies for tumor genomic profiling. J. Clin. Oncol. 2013, 31, 1815–1824.
- Chang, K.; Creighton, C.J.; Davis, C.; Donehower, L.; Drummond, J.; Wheeler, D.; Ally, A.; Balasundaram, M.; Birol, I.; Butterfield, Y.S.N.; et al. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120.
- Janiszewska, M. The microcosmos of intratumor heterogeneity: The space-time of cancer evolution. Oncogene 2019, 1–9.
- Zhang, J.; Fujimoto, J.; Zhang, J.; Wedge, D.C.; Song, X.; Zhang, J.; Seth, S.; Chow, C.-W.; Cao, Y.; Gumbs, C.; et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014, 346, 256–259.
- Gerlinger, M.; Horswell, S.; Larkin, J.; Rowan, A.J.; Salm, M.P.; Varela, I.; Fisher, R.; McGranahan, N.; Matthews, N.; Santos, C.R.; et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat. Genet. 2014, 46, 225–233.
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892.
- Prasad, V. Perspective: The precision-oncology illusion. Nature 2016, 537, S63.
- Rodon, J.; Soria, J.-C.; Berger, R.; Miller, W.H.; Rubin, E.; Kugel, A.; Tsimberidou, A.; Saintigny, P.; Ackerstein, A.; Braña, I.; et al. Genomic and transcriptomic profiling expands precision cancer medicine: The WINTHER trial. Nat. Med. 2019, 25, 751–758.
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765.
- Soeda, H.; Shimodaira, H.; Gamoh, M.; Ando, H.; Isobe, H.; Suto, T.; Takahashi, S.; Kakudo, Y.; Amagai, K.; Mori, T.; et al. Phase II trial of cetuximab plus irinotecan for oxaliplatin- and irinotecan-based chemotherapy-refractory patients with advanced and/or metastatic colorectal cancer: Evaluation of efficacy and safety based on KRAS mutation status (T-CORE0801). Oncology 2014, 87, 7–20.
- Amado, R.G.; Wolf, M.; Peeters, M.; Cutsem, E.V.; Siena, S.; Freeman, D.J.; Juan, T.; Sikorski, R.; Suggs, S.; Radinsky, R.; et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 1626–1634.
- Bokemeyer, C.; Cutsem, E.V.; Rougier, P.; Ciardiello, F.; Heeger, S.; Schlichting, M.; Celik, I.; Köhne, C.-H. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: Pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur. J. Cancer 2012, 48, 1466–1475.
- Price, T.J.; Peeters, M.; Kim, T.W.; Li, J.; Cascinu, S.; Ruff, P.; Suresh, A.S.; Thomas, A.; Tjulandin, S.; Zhang, K.; et al. Panitumumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer (ASPECCT): A randomised, multicentre, open-label, non-Inferiority phase 3 study. Lancet Oncol. 2014, 15, 569–579.
- Ye, L.-C.; Liu, T.-S.; Ren, L.; Wei, Y.; Zhu, D.-X.; Zai, S.-Y.; Ye, Q.-H.; Yu, Y.; Xu, B.; Qin, X.-Y.; et al. Randomized controlled trial of cetuximab plus chemotherapy for patients with KRAS wild-type unresectable colorectal liver-limited metastases. J. Clin. Oncol. 2013, 31, 1931–1938.
- Douillard, J.-Y.; Siena, S.; Cassidy, J.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Randomized phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: The PRIME Study. J. Clin. Oncol. 2010, 28, 4697–4705.
- Arnold, D.; Lueza, B.; Douillard, J.-Y.; Peeters, M.; Lenz, H.-J.; Venook, A.; Heinemann, V.; Cutsem, E.V.; Pignon, J.-P.; Tabernero, J.; et al. Prognostic and predictive value of primary tumour side in patients with RAS wild-type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomized trials. Ann. Oncol. 2017, 28, 1713–1729.
- García-Foncillas, J.; Sunakawa, Y.; Aderka, D.; Wainberg, Z.; Ronga, P.; Witzler, P.; Stintzing, S. Distinguishing features of cetuximab and panitumumab in colorectal cancer and other solid tumors. Front. Oncol. 2019, 9, 849.
- Goldberg, R.M.; Montagut, C.; Wainberg, Z.A.; Ronga, P.; Audhuy, F.; Taieb, J.; Stintzing, S.; Siena, S.; Santini, D. Optimising the use of cetuximab in the continuum of care for patients with metastatic colorectal cancer. ESMO Open 2018, 3, e000353.
- Lu, M.; Zhan, X. The crucial role of multiomic approach in cancer research and clinically relevant outcomes. EPMA J. 2018, 9, 77–102.
- Stunnenberg, H.G.; Hubner, N.C. Genomics meets proteomics: Identifying the culprits in disease. Hum. Genet. 2014, 133, 689–700.
- Manzoni, C.; Kia, D.A.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; Lewis, P.A.; Ferrari, R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Brief. Bioinform. 2016, 19, 114.
- Kumar, D.; Bansal, G.; Narang, A.; Basak, T.; Abbas, T.; Dash, D. Integrating transcriptome and proteome profiling: Strategies and applications. Proteomics 2016, 16, 2533–2544.
- Akbani, R.; Ng, P.K.S.; Werner, H.M.J.; Shahmoradgoli, M.; Zhang, F.; Ju, Z.; Liu, W.; Yang, J.-Y.; Yoshihara, K.; Li, J.; et al. A pan-cancer proteomic perspective on the cancer genome atlas. Nat. Commun. 2014, 5, 3887.
- Krug, K.; Jaehnig, E.J.; Satpathy, S.; Blumenberg, L.; Karpova, A.; Anurag, M.; Miles, G.; Mertins, P.; Geffen, Y.; Tang, L.C.; et al. Proteogenomic landscape of breast cancer tumorigenesis and targeted therapy. Cell 2020, 183, 1436–1456.
- Leong, A.S.-Y.; Zhuang, Z. The changing role of pathology in breast cancer diagnosis and treatment. Pathobiology 2011, 78, 99–114.
- Peng, B.; Li, H.; Peng, X.-X. Functional metabolomics: From biomarker discovery to metabolome reprogramming. Protein Cell 2015, 6, 628–637.
- Spratlin, J.L.; Serkova, N.J.; Eckhardt, S.G. Clinical applications of metabolomics in oncology: A review. Clin. Cancer Res. 2009, 15, 431–440.
- Gowda, G.N.; Zhang, S.; Gu, H.; Asiago, V.; Shanaiah, N.; Raftery, D. Metabolomics-based methods for early disease diagnostics. Expert Rev. Mol. Diagn. 2014, 8, 617–633.
- Long, Y.; Sanchez-Espiridion, B.; Lin, M.; White, L.; Mishra, L.; Raju, G.S.; Kopetz, S.; Eng, C.; Hildebrandt, M.A.T.; Chang, D.W.; et al. Global and targeted serum metabolic profiling of colorectal cancer progression. Cancer 2017, 123, 4066–4074.
- Chaturvedi, A.; Cruz, M.M.A.; Jyotsana, N.; Sharma, A.; Yun, H.; Görlich, K.; Wichmann, M.; Schwarzer, A.; Preller, M.; Thol, F.; et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood 2013, 122, 2877–2887.
- Zhang, Y.; He, C.; Qiu, L.; Wang, Y.; Qin, X.; Liu, Y.; Li, Z. Serum unsaturated free fatty acids: A potential biomarker panel for early-stage detection of colorectal cancer. J. Cancer 2016, 7, 477–483.
- Ben-David, U.; Beroukhim, R.; Golub, T.R. Genomic evolution of cancer models: Perils and opportunities. Nat. Rev. Cancer 2019, 19, 97–109.
- Cekanova, M.; Rathore, K. Animal models and therapeutic molecular targets of cancer: Utility and limitations. Drug Des. Dev. Ther. 2014, 8, 1911–1922.
- Tratar, U.L.; Horvat, S.; Cemazar, M. Transgenic mouse models in cancer research. Front. Oncol. 2018, 8, 268.
- Day, C.-P.; Merlino, G.; Van Dyke, T. Preclinical mouse cancer models: A maze of opportunities and challenges. Cell 2015, 163, 39–53.
- Politi, K.; Pao, W. How genetically engineered mouse tumor models provide insights into human cancers. J. Clin. Oncol. 2011, 29, 2273–2281.
- Van Staveren, W.C.G.; Solís, D.Y.W.; Hébrant, A.; Detours, V.; Dumont, J.E.; Maenhaut, C. Human cancer cell lines: Experimental models for cancer cells in situ? For cancer stem cells? Biochim. Biophys. Acta BBA Rev. Cancer 2009, 1795, 92–103.
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Techn. 2014, 12, 207–218.
- Clevers, H.; Tuveson, D.A. Organoid models for cancer research. Annu. Rev. Cancer Biol. 2019, 3, 223–234.
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418.
- Bock, C.; Boutros, M.; Camp, J.G.; Clarke, L.; Clevers, H.; Knoblich, J.A.; Liberali, P.; Regev, A.; Rios, A.C.; Stegle, O.; et al. The organoid cell atlas. Nat. Biotechnol. 2020, 1–5.
- Schütte, M.; Risch, T.; Abdavi-Azar, N.; Boehnke, K.; Schumacher, D.; Keil, M.; Yildiriman, R.; Jandrasits, C.; Borodina, T.; Amstislavskiy, V.; et al. Molecular dissection of colorectal cancer in pre-clinical models identifies biomarkers predicting sensitivity to EGFR inhibitors. Nat. Commun. 2017, 8, 14262.
- Byrne, A.T.; Alférez, D.G.; Amant, F.; Annibali, D.; Arribas, J.; Biankin, A.V.; Bruna, A.; Budinská, E.; Caldas, C.; Chang, D.K.; et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat. Rev. Cancer 2017, 17, 254–268.
- Katt, M.E.; Placone, A.L.; Wong, A.D.; Xu, Z.S.; Searson, P.C. In vitro tumor models: Advantages, disadvantages, variables, and selecting the right platform. Front. Bioeng Biotechnol. 2016, 4, 12.
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; Jong, S.D.; Jonkers, J.; Maelandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013.
- Hynds, R.E.; Vladimirou, E.; Janes, S.M. The secret lives of cancer cell lines. Dis. Model. Mech. 2018, 11, dmm037366.
- Hughes, P.; Marshall, D.; Reid, Y.; Parkes, H.; Gelber, C. The costs of using unauthenticated, over-passaged cell lines: How much more data do we need? Biotechniques 2007, 43, 575–586.
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607.
- Ruggeri, B.A.; Camp, F.; Miknyoczki, S. Animal models of disease: Pre-clinical animal models of cancer and their applications and utility in drug discovery. Biochem. Pharmacol. 2014, 87, 150–161.
- Bibby, M.C. Orthotopic models of cancer for preclinical drug evaluation advantages and disadvantages. Eur. J. Cancer 2004, 40, 852–857.
- Huynh, A.S.; Abrahams, D.F.; Torres, M.S.; Baldwin, M.K.; Gillies, R.J.; Morse, D.L. Development of an orthotopic human pancreatic cancer xenograft model using ultrasound guided injection of cells. PLoS ONE 2011, 6, e20330.
- Eglen, R.M.; Klein, J.-L. Three-dimensional cell culture: A rapidly emerging approach to cellular science and drug discovery. SLAS Discov. 2017, 22, 453–455.
- Sittampalam, S.; Eglen, R.; Ferguson, S.; Maynes, J.T.; Olden, K.; Schrader, L.; Shelper, T.; Ferrer, M. Three-dimensional cell culture assays: Are they more predictive of in vivo efficacy than 2D monolayer cell-based assays? Assay Drug. Dev. Technol. 2015, 13, 254–261.
- Langhans, S.A. Three-dimensional in vitro cell culture models in drug discovery and drug repositioning. Front. Pharmacol. 2018, 9, 6.
- Koch, J.; Mönch, D.; Maaß, A.; Gromoll, C.; Hehr, T.; Leibold, T.; Schlitt, H.J.; Dahlke, M.-H.; Renner, P. Three dimensional cultivation increases chemo- and radioresistance of colorectal cancer cell lines. PLoS ONE 2021, 16, e0244513.
- Weeber, F.; Ooft, S.N.; Dijkstra, K.K.; Voest, E.E. Tumor organoids as a pre-clinical cancer model for drug discovery. Cell Chem. Biol. 2017, 24, 1092–1100.
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330.
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926.
- L’Espérance, S.; Bachvarova, M.; Tetu, B.; Mes-Masson, A.-M.; Bachvarov, D. Global gene expression analysis of early response to chemotherapy treatment in ovarian cancer spheroids. BMC Genom. 2008, 9, 99.
- Zietarska, M.; Maugard, C.M.; Filali-Mouhim, A.; Alam-Fahmy, M.; Tonin, P.N.; Provencher, D.M.; Mes-Masson, A. Molecular description of a 3D in vitro model for the study of epithelial ovarian cancer (EOC). Mol. Carcinogen. 2007, 46, 872–885.
- Bogdanowicz, D.R.; Lu, H.H. Studying cell-cell communication in co-culture. Biotechnol. J. 2013, 8, 395–396.
- Betriu, N.; Semino, C.E. Development of a 3D co-culture system as a cancer model using a self-assembling peptide scaffold. Gels 2018, 4, 65.
- Nii, T.; Makino, K.; Tabata, Y. Three-dimensional culture system of cancer cells combined with biomaterials for drug screening. Cancers 2020, 12, 2754.
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.-H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid modeling of the tumor immune microenvironment. Cell 2018, 175, 1972–1988.
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; Van Haar, J.D.; Fanchi, L.F.; Slagter, M.; Van Velden, D.L.D.; Kaing, S.; Kelderman, S.; et al. Generation of tumor-reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell 2018, 174, 1586–1598.
- Schnalzger, T.E.; Groot, M.H.; Zhang, C.; Mosa, M.H.; Michels, B.E.; Röder, J.; Darvishi, T.; Wels, W.S.; Farin, H.F. 3D Model for CAR-mediated cytotoxicity using patient-derived colorectal cancer organoids. EMBO J. 2019, 38.
- Nozaki, K.; Mochizuki, W.; Matsumoto, Y.; Matsumoto, T.; Fukuda, M.; Mizutani, T.; Watanabe, M.; Nakamura, T. Co-culture with intestinal epithelial organoids allows efficient expansion and motility analysis of intraepithelial lymphocytes. J. Gastroenterol. 2016, 51, 206–213.
- Bar-Ephraim, Y.E.; Kretzschmar, K.; Clevers, H. Organoids in immunological research. Nat. Rev. Immunol. 2020, 20, 279–293.
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34.
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028.
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung CANCER. N. Engl. J. Med. 2015, 373, 1627–1639.
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Cutsem, E.V.; McDermott, R.; Hill, A.; et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J. Clin. Oncol. 2018, 36, 773–779.
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.-J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet. Oncol. 2017, 18, 1182–1191.
- Klein, E.; Hau, A.-C.; Oudin, A.; Golebiewska, A.; Niclou, S.P. Glioblastoma organoids: Pre-clinical applications and challenges in the context of immunotherapy. Front. Oncol. 2020, 10, 604121.
- Mertz, D.R.; Ahmed, T.; Takayama, S. Engineering cell heterogeneity into organs-on-a-chip. Lab Chip 2018, 18, 2378–2395.
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662–1668.
- Jeong, S.-Y.; Lee, J.-H.; Shin, Y.; Chung, S.; Kuh, H.-J. Co-culture of tumor spheroids and fibroblasts in a collagen matrix-incorporated microfluidic chip mimics reciprocal activation in solid tumor microenvironment. PLoS ONE 2016, 11, e0159013.
- Wnorowski, A.; Yang, H.; Wu, J.C. Progress, obstacles, and limitations in the use of stem cells in organ-on-a-chip models. Adv. Drug Deliver. Rev. 2018.
- Skardal, A.; Devarasetty, M.; Forsythe, S.; Atala, A.; Soker, S. A reductionist metastasis-on-a-chip platform for in vitro tumor progression modeling and drug screening. Biotechnol. Bioeng. 2016, 113, 2020–2032.
- Skardal, A.; Shupe, T.; Atala, A. Organoid-on-a-chip and body-on-a-chip systems for drug screening and disease modeling. Drug Discov. Today 2016, 21, 1399–1411.