Autism spectrum disorder (ASD) encompasses a spectrum of early-onset neurodevelopmental disorders with an estimated prevalence of ~1.5% in developed countries. Patients present early deficits in social interaction and communication, repetitive patterns of behavior, and restricted interests and activities.Chromodomain helicase domain 8 (CHD8) is one of the most frequently mutated and most penetrant genes in the autism spectrum disorder (ASD). Individuals with CHD8 mutations show leading symptoms of autism, macrocephaly, and facial dysmorphisms. The molecular and cellular mechanisms underpinning the early onset and development of these symptoms are still poorly understood and prevent timely and more efficient therapies of patients.
- ASD
- CHD8
- single-cell sequencing
- excitatory/inhibitory imbalance
1. A Role of CHD8 in Gene Regulation
The linear DNA-strand of eukaryotic cells is coiled around two copies each of the core histone proteins H2A, H2B, H3 and H4 to form a nucleosome, the basic unit of the chromatin. Further coiling of the chromatinized DNA-strand in 3D-space leads to the formation of a closed, transcriptionally inactive, chromatin structure. Epigenetic processes, such as posttranslational modifications of free histone tails, chromatin remodeling, and DNA modifications, act together to regulate chromatin conformation in a spatially and temporally controlled manner during development and beyond (reviewed in [1][2]. Consistent with this critical function, alterations in epigenetic factors driving these processes have been increasingly recognized as the genetic cause for various neurodevelopmental syndromes (reviewed in [3][4]). For instance, the chromatin remodeler (CR) CHD8 has been identified as the genetic cause of a distinct neurodevelopmental syndrome (see the following section).
CRs regulate nucleosome sliding, conformational changes of nucleosomal DNA, and exchange of histone variants. All these processes affect the access of transcription factors (TFs) to their binding sites, and consequently, gene expression (reviewed in [5]). CHD8 belongs to a subfamily of CRs that utilize ATP (adenosine triphosphate) hydrolysis to promote translocation down the DNA minor groove (reviewed in [6]). Additional domains next to the central ATPase domain are important to chromatin binding, interaction with specific histone modifications, and/or regulation of ATPase activity. In this regard, CHD8 contains an N-terminal tandem chromodomain mediating binding to methylated lysine residues in free histone tails and a C-terminal SANT-like domain supporting an association with histone tails. In addition, the C-terminus harbors a BRK domain also found in related CHD subfamilies (Figure 1).
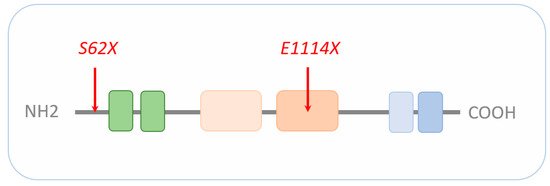
Figure 1. Schematic representation of CHD8. The signature motif of the entire CHD family is an N-terminal tandem chromodomain (green boxes) responsible for chromatin binding. The central SNF2-family ATPase domain consists of two lobes (light beige and beige box), with each containing two tandem RecA-like folds parts known as DExx and HELIC. The ATPase domain uses ATP hydrolysis to guide toward translocation down the DNA minor groove. The C-terminus contains functional motifs, such as SANT (light blue) or BRK (blue) domains. SANT domains support association with histone tails, while the BRK domain is also found in several SWI/SNF complexes. The localization of the CHD8 loss-of-function mutations S62X and E1114X are highlighted by red arrows. Drawing is not to scale and refers to the long form of CHD8. Schematic adapted from [7], attribution CC BY.
This modular structure and combinatorial use of regulatory domains within the CHD family suggests common and subtype-specific roles in chromatin remodeling (discussed in [8]). In general, CHDs, including CHD8, recognize chromatin signatures that can undergo dynamic changes, thus modulating their regulatory space. Such plasticity makes it more difficult to define genuine CHD target genes when compared to genes targeted by DNA sequence-specific TFs [9][10].
2. Disruptive de Novo Mutations of CHD8 Cause Autism and Macrocephaly
Whole exome sequencing (WES) [11][12][13][14] and targeted resequencing [15][16][17][13][18][19][20] of parent–child trios/quads have established CHD8 as one of the most frequently mutated and most penetrant genes in ASD. Heterozygote de novo mutations included frameshift, nonsense, missense, translocation, single nucleic acid deletion, and splice site variant mutations; most of these were predicted to cause a loss of function. Mutations in CHD8 were most frequently associated with autism, followed by macrocephaly [9][18][20], a phenotype also observed in patients carrying a balanced translocation disrupting CHD8 [21][22]. Orbital overgrowth developed within the first 2 months postnatally, indicating a neurodevelopmental origin, and concurred with increased head growth throughout early childhood. Patients with CHD8 mutations also represent facial dysmorphisms (prominent forehead and eyes and posteriorly rotated ears) and, to a lesser degree, recurrent obstipation and sleep disturbances. A body of recent studies further supports that disruptive de novo mutations of CHD8 underpin a neurodevelopmental syndrome with the leading symptoms of autism and macrocephaly [23][24][25][26]. More specifically, Beighley et al. [24] showed that individuals with ASD carrying CHD8 mutations display less severe adaptive deficits in communication skills and lower seizure prevalence when compared to individuals with ASD carrying other high-risk mutations. This finding suggests that a more nuanced picture of behavioral and cognitive phenotypes in individuals with CHD8 mutations is important to guide future preclinical and basic research.
3. The Excitation/Inhibition Hypothesis in ASD
Rubenstein and Merzenich [27] originally hypothesized deregulation of the balance between excitation and inhibition (E/I balance) in cortical and subcortical circuits as a key mechanism in patients with ASD. Different homeostatic and developmental processes are known to maintain the E/I balance during development and beyond (reviewed in [28]). At the level of single neurons, information processing critically depends on the balance between excitatory and inhibitory inputs and is spatially regulated by various processes, including intrinsic neuronal excitability, synaptic transmission, and homeostatic plasticity ([29], reviewed in [30]). At the circuit level, the E/I balance reflects the complex interaction between glutamatergic excitatory and GABAergic (γ-aminobutyric acid) inhibitory neurons, excitatory and inhibitory synapse formation, and overall neuronal network activity [31][32]. In the following, we will consider three independent research lines that have provided complementary evidence for the E/I balance hypothesis in ASD.
First, functional brain studies support the E/I imbalance hypothesis in patients with ASD ([33]; reviewed in [34]). For example, γ-band electrophysiological activity (30–100 Hz), a presumed proxy to the E/I balance within local neuronal circuits, is altered in patients with ASD, particularly in relation to auditory-related γ-band activity [35][36]. In addition, magnetic resonance spectroscopy (MRS) has enabled the measurement in vivo of the abundance of the excitatory and inhibitory neurotransmitters glutamate and GABA, respectively. This more direct approach corroborated alterations in neurotransmitter levels within different cortical and subcortical regions in patients with ASD [37][38][39][40]. For example, glutamate levels in the striatum were reported to correlate negatively with social impairment [38], suggesting that the E/I balance hypothesis is of clinical relevance. In light of the significant heterogeneity in ASD, patient stratification is needed to achieve better granularity in functional phenotyping.
Second, integrated functional genomic analysis has been used to narrow down specific molecular pathways and circuits in ASD. Parikshak et al. [41] mapped ASD and ID risk genes onto coexpression networks representing developmental trajectories and transcriptional profiles derived from fetal and adult cortical laminae. Notably, multiple modules enriched in ASD risk were connected by this approach to mid fetal glutamatergic projection neurons in the upper (L2/L3) and lower (L5/L6) cortical layers. In a contemporary study, Willsey et al. [42] sought to identify periods, brain regions, and cell types in which nine high confidence ASD risk genes, including CHD8, converge. Coexpression networks inferred from these “seed” genes highlighted multiple brain regions, such as the cortex, across human development and into adulthood. Assessing enrichment of an independent set of probable ASD genes enabled narrowing down a key point of convergence in mid fetal deep (L5/L6) layer cortical glutamatergic projection neurons (Figure 2).
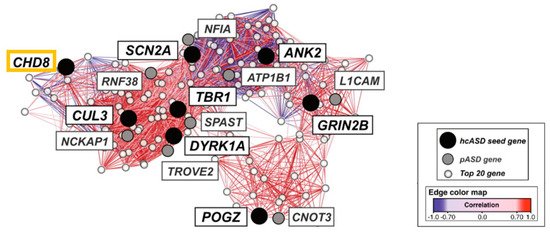
Figure 2. Mid fetal spatio-temporal coexpression network in the human prefrontal cortex. The analysis comprised weeks 13 to 19 post conception. High confidence ASD risk genes (hcASD seed gene) are marked in black with CHD8 boxed in orange. Probable ASD risk genes (pASD gene) are shown in grey, and the top 20 genes (Top 20 gene) best correlated with each hcASD gene in white. The lines (edges) represent coexpression correlations ≥0.7; positive correlations are shown in red and negative correlations are shown in blue. Graphic adapted from [42], attribution license 5004710196206.
The differences in the assignment of the specific cortical layers between these two studies [41][42] are unsurprising given the differences in the input genes selected, in the approaches to network construction, and in the selection of expression data. Collectively, these findings implicate CHD8 in glutamatergic projecting regions, which play an important role in social behavior and intellectual ability, among other higher brain functions.
Most recently, imaging genomics has highlighted genes that covary with resting-state functional MRI (rs-fMRI) measurements across cortical projecting regions (reviewed in [43]), indicating that gene expression might underpin functional signals in the human brain. To expand this approach to ASD, Berto et al. [44] integrated brain gene expression datasets of neurotypical controls and individuals with ASD and regionally matched brain activity measurements from fMRI datasets. This enabled the identification of genes linked with brain activity, whose association was disrupted in individuals with ASD. A subset of these genes showed a differential developmental trajectory in individuals with ASD relative to controls. These genes were enriched in voltage-gated ion channels and inhibitory neurons, supporting the E/I imbalance in ASD.
Third, independent support for the E/I imbalance hypothesis has grown from transgenic mice studies. Platt et al. [45] used CRISPR/Cas9 editing to produce a germline Chd8 heterozygote frameshift mutation in mice. Notably, these animals showed impaired Wingless (Wnt) signaling in the nucleus accumbens (NAs), which, together with deregulated cell adhesion, led to a reduction in local inhibitory signaling of medium spiny neurons (MSNs). Consequently, MSNs displayed an increase in spontaneous excitatory output associated with mild deficits in social interaction, elevated anxiety, and increased motor learning. In a subsequent study, Jung et al. [46] produced germline Chd8 heterozygote mice (Asn2373LysfsX2) recapitulating a mutation in patients with ASD (Asn2371LysfsX). Interestingly, hippocampal neuronal activity was suppressed in females under resting conditions and rose to neural activity of wild type mice following exposure to an environmental stressor. At the opposite, heterozygote male mice displayed normal resting neuronal activity but an enhanced response following stress exposure. Consistent with this finding, the synaptic inhibitory transmission was enhanced in female hippocampi but reduced in male hippocampi. Behavioral phenotyping of germline heterozygote Chd8 mice showed male preponderant abnormalities of social communication in pups, anxiety-like mother seeking/attachment behavior in juveniles, and isolation-induced self-grooming in adults. Taken together, this study showed that germline Chd8 heterozygosity associates with sex-preponderant behavioral deficits and sexually dimorphic inhibitory synaptic transmission in the hippocampus, a brain region strongly connected to different brain regions involved in ASD. In a most recent study, Ellingford et al. [47] investigated germline heterozygote Chd8-floxed mice that were mated to Cre-mice in which expression of Cre was directed to either all cell types (β-actin-Cre+/−) or specifically to glutamatergic (NKx.1-Cre+/−) or GABAergic (NEX-Cre+/−) neurons. Synaptic development of prefrontal pyramidal neurons was affected in a stage-specific and cell-autonomous manner in germline Cdh8 heterozygote mice and caused contrasting changes in excitatory and inhibitory synaptic transmission. Unexpectedly, heterozygote neurons responded with increased, rather than decreased, inhibitory transmission to a blockade of spontaneous transmission in ex-vivo brain slices. This inhibitory response points to the presence of dysregulated mechanisms of homeostatic plasticity in germline Chd8 heterozygote prefrontal neurons.
In summation, results from germline Chd8 heterozygote mice strengthen the E/I balance hypothesis of ASD in general and point to sex-, stage-, tissue- and cell-type-specific abnormalities in synaptic transmission and homeostatic plasticity. While it is tempting to extrapolate from these findings to humans, more detailed insight into the molecular mechanisms driving the E/I imbalance in mice and human models of brain development are needed.
4. Single-Cell Sequencing of Brain Cells with CHD8 Mutations
4.1. Defining the Molecular Identity of Neural Cells
Multipotent cells of any organ, including the human brain, develop and differentiate along specific lineage trajectories to produce distinct cell types whose well-function is critical to sustaining human health. Cellular mechanisms underpinning the onset and course of neurodevelopmental disorders are still poorly understood, thus limiting timely and more efficient therapies of ASD. Progress in this area will require an understanding of “when, why and how cells deviate from their normal trajectories” [48] (p. 377). To realize this ambitious goal, it will be necessary to establish a detailed molecular roadmap of cellular development across space and time with sufficient resolution. High-throughput single-cell sequencing technologies directly quantify information-bearing molecules, such as DNA and/or RNA, that encode and enact each cell’s biological identity. Fine-grained, broad-scope single-cell sequencing data can advance our understanding of the identity of hundreds of cellular phenotypes that form the building blocks of neural circuits (reviewed in [49]). Beyond that, single-cell sequencing of cells from in vivo and/or in vitro models of neurodevelopment can inform us “when, why and how” the presence of high confidence risk genes interferes with normal brain development in ASD.
4.2. Single-Cell Sequencing of CHD8 Knockout during Mouse Neocorticogenesis
The combination of single-cell RNA sequencing (sc-RNAseq) and CRISPR/Cas9 technologies, called Perturb-Seq, allows the production of information-rich transcriptomes and to introduce and analyze at the same time the effects of genetic perturbations. In CRISPR/Cas9, a guide RNA (gRNA) is designed to bind to the DNA sequence that is to be edited. The Cas9 enzyme then binds to the gRNA and induces a break in the DNA. The cell often incorrectly repairs this break and produces a gene “knockout” with a partial or entire loss of protein function. In Perturb-Seq, each cell is then sequenced independently so that different genetic manipulations and numerous genes can be investigated in one experiment across all cell types and states.
Based on this approach, Jin et al. [50] recently sought to address the role of a panel of high-risk ASD/NDD genes, including CHD8, during moue corticogenesis in vivo (Figure 3)
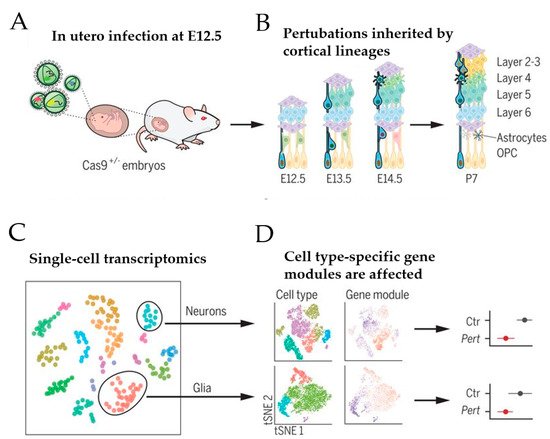
Figure 3. Schematic of in vivo Perturb-Seq analysis of mouse corticogenesis. (A) The lateral ventricles of Cas9+/− embryos were infected at E12.5 with lentiviral particles that contained a lentiviral guide RNA (gRNA) library targeting ASD/NDD risk genes. (B) ASD/NDD risk genes were gene-edited (“knocked-out”) in infected neural progenitor cells. These mutations were passed on to their progeny, including cells of the cortical lineage that form the upper and lower layer of the cortex. (C) At postnatal day 7, cortices were dissected and used for single-cell sequencing. (D) Gene expression modules were affected in a manner dependent on the individual gene perturbation and the specific cell type. Scheme adapted from [50], attribution license 5004760017864.
A transgenic mouse line that constitutively expressed Cas9 was chosen to infect the developing lateral ventricles of Cas9 heterozygote embryos in utero with pools of gRNAs, each targeting a specific high-risk ASD/NDD gene (Figure 3A). To enhance knockout efficiency, each lentiviral vector harbored two different gRNAs complementary to the coding exons of one ASD/NDD gene and a blue fluorescent protein (BFP) reporter with a distinct barcode corresponding to the perturbation target. The lentiviral injection was carried out on embryonic day 12.5, leading to the infection of neural progenitors lining the lateral ventricle of the developing neocortex and the ganglionic eminence. This approach enabled investigation of the effect of each perturbation across a wide range of cell types from distinct brain regions, including cortical projection neurons, interneurons, astroglia, and oligodendrocytes (Figure 3B). Under sparse labeling conditions, less than 0.1% of cells in the cortex were infected, implicating that these cells developed amidst unperturbed neighboring cells. At postnatal day 7, infected cortical cells were isolated by fluorescence-activated cell sorting (FACS) and used in a droplet-based scRNA-seq (see [49] for an introduction to this technique) to assess each cell’s expression profile along with its perturbation code. This analysis revealed that 40 to 70% of the FACS-positive cells contained frameshift insertion or deletion for each gRNA target. After filtering out low-quality cells, 35,847 cells remained that comprised cortical projection neurons, cortical inhibitory neurons, astrocytes, oligodendrocytes, and microglia (Figure 3C). Among these cells, 50% carried barcodes for a single gene perturbation with a median of 338 cells per perturbation. Together, these perturbations embraced 35 ASD/NDD risk genes, including Chd8. Since the small number of cells for any given perturbation per cell type prevented differential expression analysis between gene-edited and normal cells, Jin et al. [50] calculated the effect size of each perturbation on correlated expression modules across cell types compared with cells infected with control vector (i.e., lentiviral vector expressing green fluorescence protein) (Figure 3D).
Interestingly, perturbation of Chd8 affected the expression of a gene module highly expressed in oligodendrocyte precursor cells (OPCs) but lowly in committed oligodendrocyte progenitors (COPs) and newly formed oligodendrocytes (NFOLs). This finding suggested a role of Chd8 in the development of the oligodendrocyte lineage, a hypothesis further supported by in-situ hybridization and immunohistochemistry analysis of marker genes (Cspg4, Pdgfra, and MBP) in cortices from Chd8 germline heterozygote mice. In agreement with these findings, previous studies on Chd8 germline heterozygote mice [51][52] have found that combinatorial interactions of Chd8 with lineage-specific TFs (e.g., Olig2), chromatin modifiers (e.g., KMT2), and chromatin remodelers (e.g., CHD7) coordinate the temporal and spatial control of oligodendrocyte lineage-specific gene regulation. Chd8 heterozygosity is associated with early proliferation defects of OPCs, impaired oligodendrocyte differentiation, and circumscribed myelination defects. Notably, cognitive, behavioral, and motor deficits in patients with ASD have been attributed to myelination defects, including frequent central white matter abnormalities consisting of deficits in myelin content and compaction [53][54]. Likewise, one-third of the individuals with a mutation in CHD8 present variable degrees of ventriculomegaly and delayed myelination [55]. Taken together, these findings indicate that disruption of Chd8 during mid fetal neocorticogenesis in mice elicits postnatal perturbations in gene expression modules relevant to clinical phenotypes.
Unexpectedly though, Chd8 editing during mid fetal neocorticogenesis did not perturb gene modules related to excitatory and/or inhibitory neurons. This result is in stark contrast to those from previous studies on germline heterozygote Chd8 miceand from integrated functional genomics of ASD/NDD risk genes [41][42]. We consider further possible explanations for these discrepancies in the discussion section.
4.3. Organoids as a Model for Human Brain Development
Embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) represent powerful tools to model in vitro early steps of human neurodevelopment. These cells can be differentiated in virtually any cell type relevant to ASD and have eased restraints from the limited availability of embryonic brain tissue and from postmortem brain tissue to inform on early disease processes. Furthermore, ESC/iPSC derived cells are not inflicted by secondary alterations owing to disease course, therapy, or patients’ life history and provide unlimited access to cell populations ranging from neural stem cells (NSCs) to neural progenitor cells (NPCs) to neurons (reviewed in [56]). However, neuronal differentiation in monolayer culture does not recapitulate the three-dimensional (3D) organization of the human brain and well-known structure–function relationships [57] that are important to understand the cellular origin of ASD. In this respect, pluripotent stem cell-derived brain organoids represent a tractable reductionist system that allows the modeling of molecular and physiological aspects in space and time [48].
Human brain organoids recapitulate in vitro many features of fetal brain development, including cytoarchitecture, cell diversity, and maturation (reviewed in [58]). In this respect, cerebral organoids show the progressive features of cortical development, starting with the formation of ventricular and subventricular zone-like structures and the development of organized neuronal cell layers. Cortical organoids at day 10 (D10) consist almost exclusively of Sox2-positive radial glial cells recapitulating the developing human cortical ventricular zone. These multipotent progenitors give rise to increasing proportions of intermediate progenitors (TBR2 positive) and lower layer (CTIP positive), and upper layer (SATB2 positive) cortical neurons.
Among mature neurons, glutamatergic neurons are the most abundant cell type, while GABAergic interneurons are few. Glial cells are at least equally abundant as neurons and contribute to neuronal activity, synaptogenesis, and circuit remodeling. OPCs have also been detected in cortical organoids, where they support partial myelination of axons. Mature oligodendrocytes develop only under long-term culture and then do not implement the complex organization of myelin sheets, which is necessary to enhance the propagation of nerve impulses.
A meta-analysis of single-cell transcriptomes supports that cerebral organoids recapitulate gene expression programs of in vivo neocorticogenesis up to 24 weeks after conception [59]. The long-term culture of cerebral organoids is compromised by increasing tissue heterogeneity [60] and necrosis in the organoid core [61]. In the absence of functional vascularization, insufficient oxygenation and nutrient diffusion remain severe bottlenecks that curtail later maturation stages. These bottlenecks might also contribute to high organoid-to-organoid variability that has raised concerns about the consistency of developmental processes outside the context of human embryogenesis [60]. On the other hand, Velasco et al. [62] recently showed that patterned forebrain organoids exhibit a nearly indistinguishable repertoire of cell types when compared to human brains and trace developmental trajectories with similar variability.
Overall, organoids represent a tractable entry point for studying the role of ASD risk genes in early brain development [58].
4.4. Bulk Sequencing of CHD8 Knockout Cerebral Organoids
In 2017, Wang et al. [63] first carried out gene expression profiling on tissue homogenate from CHD8 heterozygote cerebral organoids. Although this approach did not apply sc-RNAseq, we nevertheless chose to include this work given its relevance to the E/I imbalance hypothesis. Wang et al. utilized a previously generated isogenic pair of iPSCs from a healthy male donor in which one copy of CHD8 had been edited via CRISPR/Cas9 to produce an N-terminal truncation [64].
Bulk-sequencing of cerebral organoids aged 50 days revealed gene expression profiles resembling the first-trimester telencephalon. Among 559 differentially expressed genes (DEGs), 288 were up- and 271 were downregulated in CHD8 heterozygote cerebral organoids, and 203 were predicted to contain a CHD8 bindings site in their promoter region. DEGs included TCF4 (a basic helix-loop-helix TF), POU3F2 (a member of the POU family of TFs), and AUTS2 (a chromatin-remodeling factor that acts in the context of the Polycomb repressive complex 1 (reviewed in [65]); all three factors have also been implicated in schizophrenia and bipolar disorders. Analysis of enriched pathways and disease association suggested that CHD8 directly or indirectly controlled critical aspects of brain development, including neurogenesis, neuronal differentiation, forebrain development, axonal guidance and wingless/β-catenin signaling.
Interestingly, two of the three top-ranked DEGs in CHD8 heterozygote cerebral organoids were DLX6-AS1 (distal-less homeobox antisense 1) and DLX1 (distal less homeobox 1) that were upregulated ~39- and ~13-fold, respectively. Members of the DLX gene family contain a homeobox that is related to that of Distal-less (Dll), a gene expressed in the head and limbs of the developing fruit fly. Likewise, the six members of the mammalian Dlx gene family are expressed in the nervous system, neural crest derivatives, branchial arches, and developing appendages and have been implicated in patterning and development of the brain, craniofacial structures, and the axial and appendicular skeleton (reviewed in [66]). In mice, a splice variant of Dlx6-AS1, called Dlx6-AS2, cooperates with Dlx1 and Dlx2 proteins in coactivation of the Dlx5/Dlx6 enhancer [67] and promotes differentiation of GABAergic interneurons in the developing forebrain [68][69]. Consistent with this scenario, genes with a role in cerebral GABAergic interneuron differentiation, including FEZF2, ARX, and CNTN2, were differentially expressed in CHD8 heterozygote organoids.
Independent support of the finding by Wang et al. [64] is provided by a prior study by Mariani et al. [70], who detected as well upregulation of DLX6-AS1 among top-ranked DEGs. In this RNA-seq study, iPSCs were derived from four families, each of which included an individual with idiopathic ASD and increased head circumference. Importantly, GABAergic neurons were overproduced in cerebral organoids derived from individuals with idiopathic autism due to an increase in FOXG1 gene expression, a key regulator of forebrain development.
Taken together, these two studies suggest that a shift toward GABAergic neuron fate is common to both individuals with CHD8 mutations and idiopathic autism. Future studies are needed to uncover through which mechanisms CHD8 and FOXG1 upregulate DLX6-AS1 expression.
4.5. Single-Cell Sequencing of CHD8 Knockout Cerebral Organoids
Most recently, Villa et al. [71] carried out sc-RNAseq of human cerebral organoids to assess the effect of various CHD8 mutations on human neocorticogenesis. CRISPR/Cas9 editing in a human ESC line was used to delete either the C-terminal helicase domain or introduce two patient-specific mutations, each resulting in a premature stop codon (i.e., S62X and E1114X; see Figure 1). Notably, both mutations are associated with autism; yet, only the E1114X mutation is associated additionally with macrocephaly and ID [9][26], suggesting differences in etiopathogenesis.
Addressing previous concerns on organoid-to-organoid variability, the researchers first optimized culture conditions so that more than 90% of the embryo bodies successfully differentiated into cerebral organoids. At D20, CHD8 mutant organoids were either equally large or slightly smaller than their wild type counterparts. Thereafter, CHD8 mutant organoids, except those containing the S62X mutation, gained in size and surpassed wild type organoids by ~50% at D120. This differential growth pattern suggested that patient mutations associated with macrocephaly drove organoid overgrowth, while the patient mutation unassociated with macrocephaly preserved normal organoid growth.
Droplet-based scRNAseq of three different developmental stages (i.e., D20, D60, and D120) evidenced 10 different cell populations, including three clusters of radial glial cells, intermediate progenitors, interneuron progenitors (IN-IPs), interneurons (INs), early excitatory neurons, and excitatory neurons of upper (EN1) and lower (EN2) layers. Interestingly, subsequent analysis of the relative densities of these cell populations in wild type and oversized CHD8 heterozygote organoids showed stage-specific alterations, including an advanced production of IN and IN-IP and a delayed production of EN1-EN2. Consistent with this finding, temporal analysis of developmental branches supported an overrepresentation of CHD8 heterozygote cells in the interneuron branch at D60 and in the excitatory branch at D120. Among changes in cell populations due to CHD8 heterozygosity, the increase in interneurons of the parvalbumin lineage at D60 represented the most robust shift.
Further immunohistochemistry and cell proliferation studies suggested that this shift in cell populations originated from a proliferative imbalance of neural progenitors that led to an expansion of this compartment and in the number of later neurons. Concurrently though, the number of Tbr2-positive intermediate progenitor decreased and led to a reduced thickness of later cortical layers. This differential shift occurred in a cell-autonomous manner as evidenced from mosaic organoids containing wild type and CHD8 heterozygote cells in equal parts.
Bulk sequencing identified 868 DEGs at D10, among which equal amounts were up- or downregulated in CHD8 heterozygote organoids and were significantly enriched in high confidence ASD genes, such as the chromatin regulator ASHL1. In agreement with previous experimental studies on CHD8 loss of function mutations (reviewed in [10][72]), upregulated genes were enriched for those involved in cell cycle progression, RNA splicing, and transcription, while downregulated genes were enriched for those involved in neuronal differentiation and brain development. Among upregulated genes, 47% were predicted to be CHD8-bound, suggesting direct regulation, whereas, among the downregulated genes, only 28% were predicted to be CH8-bound, suggesting indirect regulation.
Moreover, stage-specific sc-RNAseq analysis corroborated the division between proliferation and neurogenesis enrichments from bulk sequencing and showed that up- and downregulated genes mapped onto DEGs in different D20 cell clusters. Interestingly, upregulated genes related to cell cycle, mRNA metabolism, ribosome biogenesis, and translation initiation were enriched in clusters of radial glial cells, whereas downregulated were enriched in clusters from the differentiation path. For example, ZEB2 (zinc finger E-box binding homeobox2) was strongly downregulated in CHD8 heterozygote radial glial cell. ZEB2 encodes a DNA-binding transcriptional repressor that interacts with activated SMADs, the transducers of TGF-β signaling, and with the nucleosome remodeling and histone deacetylation (NURD) complex (for the role of NURD in brain development see [8]). In particular, ZEB2 promotes the neuroepithelial differentiation into radial glial and thus presets the number of progenitors that participate in cortical expansion [73].
In summation, CHD8 heterozygote NPCs from cerebral organoids showed an imbalance between proliferation and differentiation characterized by (i) an expansion of the NPC pool with dysregulation of neuronal differentiation and cell cycle pathways in radial glial cells, (ii) a protracted proliferation of excitatory neuron progenitors leading to a reduced formation of cortical neurons from both upper and lower layers, and (iii) a robust increase in interneuron production, particularly of the parvalbumin GABAergic lineage. These deviations developed in a cell-autonomous manner, suggesting that only selected cell populations during specific developmental stages were vulnerable to CHD8 dosage.
References
- Hoffmann, A.; Ziller, M.; Spengler, D. Focus on Causality in ESC/iPSC-Based Modeling of Psychiatric Disorders. Cells 2020, 9, 366.
- Wade, A.A.; Lim, K.; Catta-Preta, R.; Nord, A.S. Common CHD8 Genomic Targets Contrast With Model-Specific Transcriptional Impacts of CHD8 Haploinsufficiency. Front. Mol. Neurosci. 2018, 11, 481.
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254.
- Tyagi, M.; Imam, N.; Verma, K.; Patel, A.K. Chromatin remodelers: We are the drivers! Nucleus 2016, 7, 388–404.
- Murgatroyd, C.; Spengler, D. Genetic variation in the epigenetic machinery and mental health. Curr. Psychiatry Rep. 2012, 14, 138–149.
- Krumm, N.; O’Roak, B.J.; Shendure, J.; Eichler, E.E. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 2014, 37, 95–105.
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304.
- Singleton, M.R.; Dillingham, M.S.; Wigley, D.B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007, 76, 23–50.
- Bernier, R.; Golzio, C.; Xiong, B.; Stessman, H.A.; Coe, B.P.; Penn, O.; Witherspoon, K.; Gerdts, J.; Baker, C.; Vulto-van Silfhout, A.T.; et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 2014, 158, 263–276.
- Barnard, R.A.; Pomaville, M.B.; O’Roak, B.J. Mutations and Modeling of the Chromatin Remodeler CHD8 Define an Emerging Autism Etiology. Front. Neurosci. 2015, 9, 477.
- Hu, Y.; Lai, Y.; Zhu, D. Transcription regulation by CHD proteins to control plant development. Front. Plant Sci. 2014, 5, 223.
- Hoffmann, A.; Spengler, D. Chromatin Remodeling Complex NuRD in Neurodevelopment and Neurodevelopmental Disorders. Front. Genet. 2019, 10, 682.
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.-H.; Narzisi, G.; Leotta, A.; et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 74, 285–299.
- Neale, B.M.; Kou, Y.; Liu, L.; Ma’ayan, A.; Samocha, K.E.; Sabo, A.; Lin, C.-F.; Stevens, C.; Wang, L.-S.; Makarov, V.; et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012, 485, 242–245.
- Gaugler, T.; Klei, L.; Sanders, S.J.; Bodea, C.A.; Goldberg, A.P.; Lee, A.B.; Mahajan, M.; Manaa, D.; Pawitan, Y.; Reichert, J.; et al. Most genetic risk for autism resides with common variation. Nat. Genet. 2014, 46, 881–885.
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444.
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.-Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23.
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250.
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241.
- O’Roak, B.J.; Stessman, H.A.; Boyle, E.A.; Witherspoon, K.T.; Martin, B.; Lee, C.; Vives, L.; Baker, C.; Hiatt, J.B.; Nickerson, D.A.; et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat. Commun. 2014, 5, 5595.
- Wang, T.; Guo, H.; Xiong, B.; Stessman, H.A.F.; Wu, H.; Coe, B.P.; Turner, T.N.; Liu, Y.; Zhao, W.; Hoekzema, K.; et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat. Commun. 2016, 7, 13316.
- Stessman, H.A.F.; Xiong, B.; Coe, B.P.; Wang, T.; Hoekzema, K.; Fenckova, M.; Kvarnung, M.; Gerdts, J.; Trinh, S.; Cosemans, N.; et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 2017, 49, 515–526.
- Talkowski, M.E.; Rosenfeld, J.A.; Blumenthal, I.; Pillalamarri, V.; Chiang, C.; Heilbut, A.; Ernst, C.; Hanscom, C.; Rossin, E.; Lindgren, A.M.; et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 2012, 149, 525–537.
- Yasin, H.; Gibson, W.T.; Langlois, S.; Stowe, R.M.; Tsang, E.S.; Lee, L.; Poon, J.; Tran, G.; Tyson, C.; Wong, C.K.; et al. A distinct neurodevelopmental syndrome with intellectual disability, autism spectrum disorder, characteristic facies, and macrocephaly is caused by defects in CHD8. J. Hum. Genet. 2019, 64, 271–280.
- An, Y.; Zhang, L.; Liu, W.; Jiang, Y.; Chen, X.; Lan, X.; Li, G.; Hang, Q.; Wang, J.; Gusella, J.F.; et al. De novo variants in the Helicase-C domain of CHD8 are associated with severe phenotypes including autism, language disability and overgrowth. Hum. Genet. 2020, 139, 499–512.
- Beighley, J.S.; Hudac, C.M.; Arnett, A.B.; Peterson, J.L.; Gerdts, J.; Wallace, A.S.; Mefford, H.C.; Hoekzema, K.; Turner, T.N.; O’Roak, B.J.; et al. Clinical Phenotypes of Carriers of Mutations in CHD8 or Its Conserved Target Genes. Biol. Psychiatry 2020, 87, 123–131.
- Ostrowski, P.J.; Zachariou, A.; Loveday, C.; Beleza-Meireles, A.; Bertoli, M.; Dean, J.; Douglas, A.G.L.; Ellis, I.; Foster, A.; Graham, J.M.; et al. The CHD8 overgrowth syndrome: A detailed evaluation of an emerging overgrowth phenotype in 27 patients. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 557–564.
- Wu, H.; Li, H.; Bai, T.; Han, L.; Ou, J.; Xun, G.; Zhang, Y.; Wang, Y.; Duan, G.; Zhao, N.; et al. Phenotype-to-genotype approach reveals head-circumference-associated genes in an autism spectrum disorder cohort. Clin. Genet. 2020, 97, 338–346.
- Sohal, V.S.; Zhang, F.; Yizhar, O.; Deisseroth, K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 2009, 459, 698–702.
- Iascone, D.M.; Li, Y.; Sümbül, U.; Doron, M.; Chen, H.; Andreu, V.; Goudy, F.; Blockus, H.; Abbott, L.F.; Segev, I.; et al. Whole-Neuron Synaptic Mapping Reveals Spatially Precise Excitatory/Inhibitory Balance Limiting Dendritic and Somatic Spiking. Neuron 2020, 106, 566–578.e8.
- Kavalali, E.T.; Monteggia, L.M. Targeting Homeostatic Synaptic Plasticity for Treatment of Mood Disorders. Neuron 2020, 106, 715–726.
- Gao, R.; Penzes, P. Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr. Mol. Med. 2015, 15, 146–167.
- Lee, E.; Lee, J.; Kim, E. Excitation/Inhibition Imbalance in Animal Models of Autism Spectrum Disorders. Biol. Psychiatry 2017, 81, 838–847.
- Oliveira, B.; Mitjans, M.; Nitsche, M.A.; Kuo, M.-F.; Ehrenreich, H. Excitation-inhibition dysbalance as predictor of autistic phenotypes. J. Psychiatr. Res. 2018, 104, 96–99.
- Port, R.G.; Oberman, L.M.; Roberts, T.P. Revisiting the excitation/inhibition imbalance hypothesis of ASD through a clinical lens. Br. J. Radiol. 2019, 92, 20180944.
- Edgar, J.C.; Fisk Iv, C.L.; Berman, J.I.; Chudnovskaya, D.; Liu, S.; Pandey, J.; Herrington, J.D.; Port, R.G.; Schultz, R.T.; Roberts, T.P.L. Auditory encoding abnormalities in children with autism spectrum disorder suggest delayed development of auditory cortex. Mol. Autism 2015, 6, 69.
- Port, R.G.; Edgar, J.C.; Ku, M.; Bloy, L.; Murray, R.; Blaskey, L.; Levy, S.E.; Roberts, T.P.L. Maturation of auditory neural processes in autism spectrum disorder—A longitudinal MEG study. Neuroimage Clin. 2016, 11, 566–577.
- Brown, M.S.; Singel, D.; Hepburn, S.; Rojas, D.C. Increased glutamate concentration in the auditory cortex of persons with autism and first-degree relatives: A (1)H-MRS study. Autism Res. 2013, 6, 1–10.
- Horder, J.; Petrinovic, M.M.; Mendez, M.A.; Bruns, A.; Takumi, T.; Spooren, W.; Barker, G.J.; Künnecke, B.; Murphy, D.G. Glutamate and GABA in autism spectrum disorder-a translational magnetic resonance spectroscopy study in man and rodent models. Transl. Psychiatry 2018, 8, 106.
- Kubas, B.; Kułak, W.; Sobaniec, W.; Tarasow, E.; Lebkowska, U.; Walecki, J. Metabolite alterations in autistic children: A 1H MR spectroscopy study. Adv. Med. Sci. 2012, 57, 152–156.
- Mescher, M.; Merkle, H.; Kirsch, J.; Garwood, M.; Gruetter, R. Simultaneous in vivo spectral editing and water suppression. NMR Biomed. 1998, 11, 266–272.
- Parikshak, N.N.; Luo, R.; Zhang, A.; Won, H.; Lowe, J.K.; Chandran, V.; Horvath, S.; Geschwind, D.H. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 2013, 155, 1008–1021.
- Willsey, A.J.; Sanders, S.J.; Li, M.; Dong, S.; Tebbenkamp, A.T.; Muhle, R.A.; Reilly, S.K.; Lin, L.; Fertuzinhos, S.; Miller, J.A.; et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 2013, 155, 997–1007.
- Bogdan, R.; Salmeron, B.J.; Carey, C.E.; Agrawal, A.; Calhoun, V.D.; Garavan, H.; Hariri, A.R.; Heinz, A.; Hill, M.N.; Holmes, A.; et al. Imaging Genetics and Genomics in Psychiatry: A Critical Review of Progress and Potential. Biol. Psychiatry 2017, 82, 165–175.
- Berto, S.; Treacher, A.; Caglayan, E.; Luo, D.; Haney, J.R.; Gandal, M.J.; Geschwind, D.H.; Montillo, A.; Konopka, G. Association between resting-state functional brain connectivity and gene expression is altered in autism spectrum disorder. medRxiv 2021.
- Platt, R.J.; Zhou, Y.; Slaymaker, I.M.; Shetty, A.S.; Weisbach, N.R.; Kim, J.-A.; Sharma, J.; Desai, M.; Sood, S.; Kempton, H.R.; et al. Chd8 mutation leads to autistic-like behaviors and impaired striatal circuits. Cell Rep. 2017, 19, 335–350.
- Jung, H.; Park, H.; Choi, Y.; Kang, H.; Lee, E.; Kweon, H.; Roh, J.D.; Ellegood, J.; Choi, W.; Kang, J.; et al. Sexually dimorphic behavior, neuronal activity, and gene expression in Chd8-mutant mice. Nat. Neurosci. 2018, 21, 1218–1228.
- Ellingford, R.A.; de Meritens, E.R.; Shaunak, R.; Naybour, L.; Basson, M.A.; Andreae, L.C. Cell-type-specific synaptic imbalance and disrupted homeostatic plasticity in cortical circuits of ASD-associated Chd8 haploinsufficient mice. bioRxiv 2020.
- Armand, E.J.; Li, J.; Xie, F.; Luo, C.; Mukamel, E.A. Single-Cell Sequencing of Brain Cell Transcriptomes and Epigenomes. Neuron 2021, 109, 11–26.
- Jin, X.; Simmons, S.K.; Guo, A.; Shetty, A.S.; Ko, M.; Nguyen, L.; Jokhi, V.; Robinson, E.; Oyler, P.; Curry, N.; et al. In vivo Perturb-Seq reveals neuronal and glial abnormalities associated with autism risk genes. Science 2020, 370, eaaz6063.
- Marie, C.; Clavairoly, A.; Frah, M.; Hmidan, H.; Yan, J.; Zhao, C.; Van Steenwinckel, J.; Daveau, R.; Zalc, B.; Hassan, B.; et al. Oligodendrocyte precursor survival and differentiation requires chromatin remodeling by Chd7 and Chd8. Proc. Natl. Acad. Sci. USA 2018, 115, E8246–E8255.
- Zhao, C.; Dong, C.; Frah, M.; Deng, Y.; Marie, C.; Zhang, F.; Xu, L.; Ma, Z.; Dong, X.; Lin, Y.; et al. Dual Requirement of CHD8 for Chromatin Landscape Establishment and Histone Methyltransferase Recruitment to Promote CNS Myelination and Repair. Dev. Cell 2018, 45, 753–768.e8.
- Hardan, A.Y.; Fung, L.K.; Frazier, T.; Berquist, S.W.; Minshew, N.J.; Keshavan, M.S.; Stanley, J.A. A proton spectroscopy study of white matter in children with autism. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 66, 48–53.
- Deoni, S.; Dean, D.; Joelson, S.; O’Regan, J.; Schneider, N. Early nutrition influences developmental myelination and cognition in infants and young children. Neuroimage 2018, 178, 649–659.
- Douzgou, S.; Liang, H.W.; Metcalfe, K.; Somarathi, S.; Tischkowitz, M.; Mohamed, W.; Kini, U.; McKee, S.; Yates, L.; Bertoli, M.; et al. The clinical presentation caused by truncating CHD8 variants. Clin. Genet. 2019, 96, 72–84.
- Ardhanareeswaran, K.; Mariani, J.; Coppola, G.; Abyzov, A.; Vaccarino, F.M. Human induced pluripotent stem cells for modelling neurodevelopmental disorders. Nat. Rev. Neurol. 2017, 13, 265–278.
- Gollo, L.L.; Roberts, J.A.; Cropley, V.L.; Di Biase, M.A.; Pantelis, C.; Zalesky, A.; Breakspear, M. Fragility and volatility of structural hubs in the human connectome. Nat. Neurosci. 2018, 21, 1107–1116.
- Chiaradia, I.; Lancaster, M.A. Brain organoids for the study of human neurobiology at the interface of in vitro and in vivo. Nat. Neurosci. 2020, 23, 1496–1508.
- Tanaka, Y.; Cakir, B.; Xiang, Y.; Sullivan, G.J.; Park, I.-H. Synthetic Analyses of Single-Cell Transcriptomes from Multiple Brain Organoids and Fetal Brain. Cell Rep. 2020, 30, 1682–1689.e3.
- Quadrato, G.; Nguyen, T.; Macosko, E.Z.; Sherwood, J.L.; Min Yang, S.; Berger, D.R.; Maria, N.; Scholvin, J.; Goldman, M.; Kinney, J.P.; et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 2017, 545, 48–53.
- Qian, X.; Su, Y.; Adam, C.D.; Deutschmann, A.U.; Pather, S.R.; Goldberg, E.M.; Su, K.; Li, S.; Lu, L.; Jacob, F.; et al. Sliced Human Cortical Organoids for Modeling Distinct Cortical Layer Formation. Cell Stem Cell 2020, 26, 766–781.e9.
- Velasco, S.; Kedaigle, A.J.; Simmons, S.K.; Nash, A.; Rocha, M.; Quadrato, G.; Paulsen, B.; Nguyen, L.; Adiconis, X.; Regev, A.; et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 2019, 570, 523–527.
- Wang, P.; Mokhtari, R.; Pedrosa, E.; Kirschenbaum, M.; Bayrak, C.; Zheng, D.; Lachman, H.M. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in cerebral organoids derived from iPS cells. Mol. Autism 2017, 8, 11.
- Wang, P.; Lin, M.; Pedrosa, E.; Hrabovsky, A.; Zhang, Z.; Guo, W.; Lachman, H.M.; Zheng, D. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in neurodevelopment. Mol. Autism 2015, 6, 55.
- Hoffmann, A.; Sportelli, V.; Ziller, M.; Spengler, D. Switch-Like Roles for Polycomb Proteins from Neurodevelopment to Neurodegeneration. Epigenomes 2017, 1, 21.
- Kraus, P.; Lufkin, T. Dlx homeobox gene control of mammalian limb and craniofacial development. Am. J. Med. Genet. A 2006, 140, 1366–1374.
- Feng, J.; Bi, C.; Clark, B.S.; Mady, R.; Shah, P.; Kohtz, J.D. The Evf-2 noncoding RNA is transcribed from the Dlx-5/6 ultraconserved region and functions as a Dlx-2 transcriptional coactivator. Genes Dev. 2006, 20, 1470–1484.
- Paina, S.; Garzotto, D.; DeMarchis, S.; Marino, M.; Moiana, A.; Conti, L.; Cattaneo, E.; Perera, M.; Corte, G.; Calautti, E.; et al. Wnt5a is a transcriptional target of Dlx homeogenes and promotes differentiation of interneuron progenitors in vitro and in vivo. J. Neurosci. 2011, 31, 2675–2687.
- Poitras, L.; Yu, M.; Lesage-Pelletier, C.; Macdonald, R.B.; Gagné, J.-P.; Hatch, G.; Kelly, I.; Hamilton, S.P.; Rubenstein, J.L.R.; Poirier, G.G.; et al. An SNP in an ultraconserved regulatory element affects Dlx5/Dlx6 regulation in the forebrain. Development 2010, 137, 3089–3097.
- Mariani, J.; Coppola, G.; Zhang, P.; Abyzov, A.; Provini, L.; Tomasini, L.; Amenduni, M.; Szekely, A.; Palejev, D.; Wilson, M.; et al. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell 2015, 162, 375–390.
- Villa, C.E.; Cheroni, C.; López-Tóbon, A.; Dotter, C.P.; Oliveira, B.; Sacco, R.; Yahya, A.C.; Morandell, J.; Gabriele, M.; Sommer, C.; et al. CHD8 haploinsufficiency alters the developmental trajectories of human excitatory and inhibitory neurons linking autism phenotypes with transient cellular defects. bioRxiv 2020.
- Hoffmann, A.; Spengler, D. Chromatin Remodeler CHD8 in Autism and Brain Development. J. Clin. Med. 2021, 10, 366.
- Benito-Kwiecinski, S.; Giandomenico, S.L.; Sutcliffe, M.; Riis, E.S.; Freire-Pritchett, P.; Kelava, I.; Wunderlich, S.; Martin, U.; Wray, G.; Lancaster, M.A. An early cell shape transition drives evolutionary expansion of the human forebrain. bioRxiv 2020.