Myeloid differentiation 88 (MyD88) is a well-established inflammatory adaptor protein.
- stroke
- pattern recognition receptors
- aneurysms
- neuro-inflammation
1. Introduction
Subarachnoid hemorrhage (SAH) is a subtype of hemorrhagic strokes and accounts for only 5% of all strokes [1]. However, the morbidity and mortality inflicted by SAH is a nightmare in this era of modern medicine as reflected by the fact that 20% of deaths occur before hospitalization and half of the patients by getting medical care die within a month [2,3]. The condition of the survivors is also not very good and almost one-third are lifelong dependent and many of the survivors with good clinical outcomes still have cognitive disorders [2]. SAHs are of concern compared to ischemic strokes as they tend to affect a younger population including those of working age group [1]. SAH accounts for almost 27% of stroke-related potential years of life lost by an individual before the age of 65 years [4]. The majority of SAH cases are due to the rupture of abnormally dilated, ballooning and weakened intracranial blood vessels at the arterial bifurcations; these abnormal rupture-prone arterial dilatations are referred to as intracranial aneurysms (ICAs) [5]. Immediately, after the rupture of ICAs, the subarachnoid space is flooded with extravasated blood with an elevation in intracranial pressure, which compromises the normal circulation of cerebrospinal fluid (CSF), ultimately leading to transient global cerebral ischemia [1,6]. The toxic effects of the blood and its degradation products further add to the injury of the brain [1,6]. There is a considerable body of evidence suggesting that during this insult, several damage-associated molecular patterns (DAMPs) are liberated from various cellular compartments, which have the capacity to upregulate inflammation after ligation of their cognizant pattern recognition receptors (PRR) [7,8,9,10,11]. Over the past few years, the concept of early brain injury and delayed brain injury has evolved to consider the events triggered immediately after the sentinel bleed up to 72 h and over 3–14 days or more, respectively [1,6,12,13]. Sterile inflammation mediates both of these injury phases and also contributes to various post-SAH complications such as cerebral vasospasm (CVS), seizures, delayed cerebral ischemia (DCI), cortical spreading depolarization (CSD), chronic hydrocephalus and infections, etc. that ultimately affect the clinical outcome of patients [9,10,14]. Therefore, local and systemic inflammation after SAH has been the target of current, intensive investigations to uncover the underlying complex mechanisms and highlight them as important drug targets.
Subarachnoid hemorrhage (SAH) is a subtype of hemorrhagic strokes and accounts for only 5% of all strokes [1]. However, the morbidity and mortality inflicted by SAH is a nightmare in this era of modern medicine as reflected by the fact that 20% of deaths occur before hospitalization and half of the patients by getting medical care die within a month [2][3]. The condition of the survivors is also not very good and almost one-third are lifelong dependent and many of the survivors with good clinical outcomes still have cognitive disorders [2]. SAHs are of concern compared to ischemic strokes as they tend to affect a younger population including those of working age group [1]. SAH accounts for almost 27% of stroke-related potential years of life lost by an individual before the age of 65 years [4]. The majority of SAH cases are due to the rupture of abnormally dilated, ballooning and weakened intracranial blood vessels at the arterial bifurcations; these abnormal rupture-prone arterial dilatations are referred to as intracranial aneurysms (ICAs) [5]. Immediately, after the rupture of ICAs, the subarachnoid space is flooded with extravasated blood with an elevation in intracranial pressure, which compromises the normal circulation of cerebrospinal fluid (CSF), ultimately leading to transient global cerebral ischemia [1][6]. The toxic effects of the blood and its degradation products further add to the injury of the brain [1][6]. There is a considerable body of evidence suggesting that during this insult, several damage-associated molecular patterns (DAMPs) are liberated from various cellular compartments, which have the capacity to upregulate inflammation after ligation of their cognizant pattern recognition receptors (PRR) [7][8][9][10][11]. Over the past few years, the concept of early brain injury and delayed brain injury has evolved to consider the events triggered immediately after the sentinel bleed up to 72 h and over 3–14 days or more, respectively [1][6][12][13]. Sterile inflammation mediates both of these injury phases and also contributes to various post-SAH complications such as cerebral vasospasm (CVS), seizures, delayed cerebral ischemia (DCI), cortical spreading depolarization (CSD), chronic hydrocephalus and infections, etc. that ultimately affect the clinical outcome of patients [9][10][14]. Therefore, local and systemic inflammation after SAH has been the target of current, intensive investigations to uncover the underlying complex mechanisms and highlight them as important drug targets.
The innate immune response relies on several germ line-encoded PRRs and among them, toll-like receptors (TLRs) are well characterized PRRs that can set into play the complex process of inflammation upon recognition of various DAMPs or pathogen-associated molecular pattern molecules (PAMPs) [15,16,17]. This inflammatory response triggers the expression of cytokines, chemokines, interferons and several other associated molecules, and at the same time, it kick starts the adaptive immune system [18,19].
The innate immune response relies on several germ line-encoded PRRs and among them, toll-like receptors (TLRs) are well characterized PRRs that can set into play the complex process of inflammation upon recognition of various DAMPs or pathogen-associated molecular pattern molecules (PAMPs) [15][16][17]. This inflammatory response triggers the expression of cytokines, chemokines, interferons and several other associated molecules, and at the same time, it kick starts the adaptive immune system [18][19].
TLRs have a key role in the stimulation of the immune system [19]. Engagement of the TLRs’ ligands transduces intracellular signals by engaging special adaptor proteins that ultimately lead to the activation and nuclear translocation of the transcription factors such as NF-κB and IRFs [19,20]. Both of these transcription factors dictate the consequences of the innate immune response activation. In humans, the TLR family is comprised of 10 members (TLR1–TLR10), while in mice there are twelve (TLR1–TLR9, TLR11–TLR13). In addition to their expression on cell surface membranes, TLRs are found in the endolysosomes, endoplasmic reticulum, lysosomes and endosomes [21]. Structurally these are composed of leucine-rich repeats and an ecto-domain, which arbitrates the PAMPs’ recognition, while a trans-membrane, and Toll/IL-1 receptor (TIR) domain instigates the downstream signaling. The ectodomain exhibits a horseshoe-like structure and through this domain TLRs can ligate their relevant PAMPs or DAMPs [22].
TLRs have a key role in the stimulation of the immune system [19]. Engagement of the TLRs’ ligands transduces intracellular signals by engaging special adaptor proteins that ultimately lead to the activation and nuclear translocation of the transcription factors such as NF-κB and IRFs [19][20]. Both of these transcription factors dictate the consequences of the innate immune response activation. In humans, the TLR family is comprised of 10 members (TLR1–TLR10), while in mice there are twelve (TLR1–TLR9, TLR11–TLR13). In addition to their expression on cell surface membranes, TLRs are found in the endolysosomes, endoplasmic reticulum, lysosomes and endosomes [21]. Structurally these are composed of leucine-rich repeats and an ecto-domain, which arbitrates the PAMPs’ recognition, while a trans-membrane, and Toll/IL-1 receptor (TIR) domain instigates the downstream signaling. The ectodomain exhibits a horseshoe-like structure and through this domain TLRs can ligate their relevant PAMPs or DAMPs [22].
2. Myeloid Differentiation Primary-Response Gene 88 (MyD88)
MyD88 is an important downstream protein member of TLR and IL-1 receptors [23]. It was originally discovered by Liebermann and Hoffman in 1990 in M1 myeloblastic leukemia cells where its expression was induced in response to the application of recombinant IL-6 or lung conditioned medium [24]. “MyD” basically stands for myeloid differentiation and “88” denotes the number in the list of induced genes [24]. It is now well established as an adaptor of inflammatory signaling pathways [23]. MyD88 is further associated with IL-1R-associated kinase (IRAK) family kinases through homotypic protein–protein interaction. Stimulation of IRAK at the cellular level initiates various responses, ultimately triggering the nuclear factor-kappa B (NFκB), mitogen-activated protein kinases (MAPK) and activator protein 1 (AP1), highlighting MyD88 as an important mediator of inflammatory pathways [25]. Except TLR3, in all other TLRs sub-stream intracellular signaling is dependent upon MyD88 [19]. Furthermore, advanced studies have revealed that the cellular pathway of the MyD88 signaling is an important mediator of sterile inflammation and it is affected by numerous associated subcellular proteins.
3. Role of TLR4-MyD88 in Post-SAH Inflammation
TLR4 is an important member of the TLR family of PRRs responsible for recognizing many pathogenic agonists including various DAMPs and PAMPs [15][16][17]. A few endogenous molecules such as fibrinogen and heme, which are released during SAH are recognized like LPS by the TLR4 [9][26]. When TLR4 is ligated, it triggers a cascade of intracellular signals that initiate the synthesis of pro-inflammatory cytokines, chemokines, and the expression of co-stimulatory molecules [16]. As sterile inflammation is an attribute of SAH, inflammation has gained importance as a target in the discovery of novel drugs against post-SAH inflammation [10]. Among TLRs, TLR4 is unique in that it can transmit downstream signaling via both the myeloid differentiation primary response protein 88 (MyD88) and the TRIF pathways to stimulate and initiate the inflammatory responses [27]. A study showed that during an early stage of SAH, neuronal inflammation and apoptosis was mediated by TLR4/MyD88-dependent and microglial-dependent pathways [28][29]. However, in the late phase of SAH, the neuronal apoptosis was mainly associated with TRIF-dependent and microglial-independent pathways [28][29]. Interestingly, the persistent upregulation of TLR4 at the brain level is linked with long term cognitive dysfunction [30]. This dual pattern of cerebral inflammation is very important because it reveals the importance of TLR4-mediated cerebral effects and proves the importance of this novel therapeutic target for the treatment of the SAH.
TLR4 is most abundantly expressed on astrocytes, neurons, microglial cells, myeloid cells, and epithelial cells in the central nervous system (CNS) [31]. Recent evidence suggests that TLR4 expression on non-neuronal and non-glial cells also contributes to the brain injury and is implicated in post-SAH complications [31]. Previously, increased levels of soluble isoforms of TLR2 and TLR4 have been observed in the cerebrospinal fluid (CSF) of SAH patients who developed hydrocephalus [32]. A recent investigation showed that in a rodent model of intraventricular haemorrhage, TLR4 mediates the CSF hypersecretion by the choroid plexus epithelium triggered in the cerebral ventricles, resulting in post-hemorrhagic hydrocephalus [33]. Moreover, results have proved that MyD88-dependent TLR4 signaling might play an important role as an inflammatory mediator on the aforementioned cell types in the CNS. Blood-borne DAMPs that bind to the TLR4 act as TLR4 ligands. Initially, DAMPs are identified by a cluster of differentiation 14 (CD-14), either in a soluble or glycosylphosphatidylinositol-anchored form, which then moves to myeloid differentiation factor 2 (MD-2) [31]. Following the ligation of TLR4 agonists, i.e., blood DAMPs, results in TLR4 oligomerization and binding to the TIRAP via TIR–TIR domain interaction as shown in
. Then, MyD88 associates with this complex. Following this association, this complex recruits further MyD88 molecules, members of the IL-1 receptor-associated family of kinases (IRAKs), and TNF receptor-associated factor 6 (TRAF6), and all of these molecules collectively give rise to the myddosome as shown in
[34].
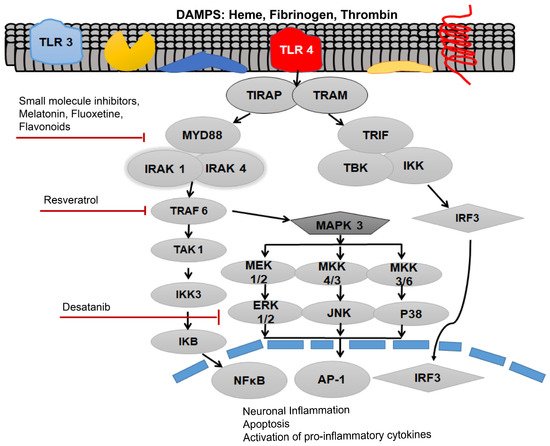
Activation of toll-like receptor 4 (TLR4) via various damage-associated molecular pattern molecules (DAMPs) to initiate downstream signaling of the receptor after subarachnoid hemorrhage (SAH). Myleoid Diffrentiation 88 (MyD88) can be inhibited by the small molecular inhibitor, melatonin, fluoxetine and various flavonoids. TNF Receptor associated factor 6 (TRAF6) can be inhibited by the Resveratrol, whereas Desatanib targets the mitogen-activated protein kinase 3 (MAPK 3) pathway. TLR4 pathway is associated with neuronal inflammation, apoptosis and activation of pro-inflammatory cytokines.
The formation of myddosome directs the downstream activation of transforming growth factor-β-activated kinase 1 (TAK1), which triggers IκB kinase (IKK) and mitogen-activated protein kinase (MAPK) signaling. The phosphorylation of IκB, leads to the activation of the nuclear translocation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), hence, stimulating the transcription of pro-inflammatory genes including various pro-inflammatory cytokines [29].
Study proves that both MyD88 and the TRIF pathways activate the expression of NF-κB, which is the foremost important transcriptional manager of inflammation-associated genes. The activation of MAPK can initiate the expression of pro-inflammatory cytokines. TNF-α can initiate vasoconstriction and oxidative stress. Furthermore, augmented TNF-α levels, especially in the brain interstitial fluid, further exacerbate cerebral vasospasm [28]. The cytokines are known to worsen the SAH-associated symptoms. IL-1β causes apoptosis, hence it further expedites the cyclooxygenase-2-associated cerebral inflammation. A downstream regulator of the MyD88, ICAM-1 is an important endothelial protein that is usually upregulated, while cerebral inflammation evidence has revealed that it might be a crucial protagonist in microcirculatory dysfunction in SAH [35]. MyD88 further activates the MAPK pathway as shown in
. MAPK and c-Jun N-terminal kinases (JNKs) signaling causes the downstream activation of activator protein 1 (AP-1), and upregulates the pro-inflammatory cytokine production [27].
MAPKs are directly associated with a lot of important cellular responses and stimuli including mitogens, heat shock, and inflammation. The MyD88-dependent pathway has also been shown to play an extremely important role in cell survival by the activation of MAPKs, such as the signal-regulated kinase (ERK), p38, and c-Jun N-terminal kinase (JNK), in response to which it activates the transcription factor activator protein-1 (AP-1) [36]. Likewise, the MAPK pathway appears to have an important role in the SAH. The MAPK pathway was a key pathway in the regulation of cerebral blood flow in the SAH induced in a rat model [37]. However, both the p38 and JNK were also found to produce neuronal and endothelial cell apoptosis, inflammatory cytokine expression, and facilitate SAH accompanied neuronal injury. In order to understand the role of MAPK pathways in post-SAH injury, recombinant osteopontin (r-OPN) was used in a rodent endovascular perforation SAH model. Pre-SAH intracerebrovascular administration of r-OPN enhanced the expression of an endogenous MAPK inhibitor, MKP-1, through interaction with L-arginyl-glycyl-L-aspartate-dependent integrin receptors and prevented vasospasm after SAH. Inhibition of this MAPK inhibitor by administering MKP-1 siRNA failed to suppress the phosphorylation of MAPKs and led to spastic cerebral arteries in a rat model at 24 h post-SAH [38]. Amazingly, it was shown that the administration of r-OPN associated increase in MAPK inhibitor (MKP-1) not only ameliorated the SAH-associated vasospasm, but also prevented from neurological impairments at 24–72 h post-SAH in a rat model.
4. MyD88 as a Therapeutic Target of Post-SAH Inflammation
Recently advanced studies have shown that the inhibition of MyD88-dependent TLR4 signaling is favorable in treating secondary injuries associated with brain hemorrhage. A few potential inhibitors of this pathway are small molecule inhibitors, aptamers, monoclonal antibodies, polyphenols and certain antibiotics.
4.1. Progesterone
Progesterone is known to modulate TLR4-MyD88-NF-
B dependent inflammation and reduces the secretion of pro-inflammatory cytokines IL-6 and TNF-α [39]. Progesterone administration has been shown to modulate the inflammation upregulated by TLR4 and downstream signaling pathways during ischemic stroke and traumatic brain injury [40]. Similarly, progesterone administration has also been shown to modulate the TLR4 and downstream pathways-dependent inflammation after SAH in rats [40]. As erstwhile explained, SAH could up-regulate expressions of both TLR4 and NF-
B. Similarly, expression of molecules downstream of TLR4/MyD88 were increased, leading to upregulation of ICAM-1, MCP-1, NF-
B, IL-1β, IL-6 and TNF-
after SAH in a rat model [40]. Progesterone administration not only downregulated the post-SAH inflammation by abrogating the expression of these inflammatory molecules in the cerebral tissue, but also improved neurological deficits, brain edema and prevented the disruption of the blood-brain barrier (BBB) [40]. Therefore, progesterone mediated pharmacological modulation of the TLR4-MyD88-NF-
B dependent inflammation after SAH requires further clinical investigation.
4.2. Small Molecule Inhibitors
Small molecule inhibitors may be used to pharmacologically target the inhibition of TLR4 and its downstream signaling involving MyD88 to curb the inflammatory response that follows SAH. Interestingly, a small molecule inhibitor of MyD88 dimerization, ST2825, has been shown to alleviate the inflammation during early brain injury after experimental SAH, and it also protects animals against SAH-induced neurological deficits [41]. Intracerebroventricular administration of ST2825 afforded the inhibition of inflammation and apoptosis by modulating the activity of several MyD88 downstream molecules such as TAK1, p38, JNK, NF-κB p65 and IκBα [41]. This study highlights the neuroprotective potential of targeting MyD88 through ST2825 during early brain injury after SAH, however, whether systemic administration of this molecule will provide similar effects to cerebroventricular administration awaits further investigation [41].
4.3. Aptamers
A novel approach to counter the inflammation after SAH is the employment of single-stranded DNA aptamers such as ApTLR#1R and ApTLR#4F. Both these aptamers block the TLR4 receptor and act as antagonizing ligands, thus inhibiting the downstream signaling via the TLR4-MyD88 pathway. Both compounds can pass the BBB, and are readily available for absorption, distribution and elimination. Moreover, these agents possess an extremely low toxicity profile. Owing to the pharmacokinetic virtues of these aptamers, rapid systemic administration, theoretically, there is no need for neurosurgical intervention for direct ventricular administration. Novel investigations are required to make these compounds stable because these are prone to degradation by nucleases. Nevertheless, these modifications may compromise the efficacy and toxicity of the aptamers [42]. Both of these aptamers are likely to be TLR4 modulators that are identified for the mitigation of post-SAH inflammation and associated complications.
4.4. Polyphenols
Resveratrol, biochanin A and curcumin are polyphenols that are isolated from plants. They can cross the BBB and significantly block the TLR4 signaling by interfering with the oligomerization of TLR-4 [43][44]. Biochanin A evidently reduces the expression levels of TLRs and its downstream signaling proteins like TIRAP, MyD88 and NF-
B pathways and the synthesis of pro-inflammatory cytokines during early brain injury after prechiasmatic blood injection in a model of SAH. Intriguingly, biochanin A administration was associated with reduced neuronal apoptosis, which was linked to improved neurological scores assessed by Garcia Scale scores, and improved spatial learning and cognitive memory assessed by modified water maze tests after SAH [45].
Resveratrol (3,4′,5-trihydroxystilbene) has been suggested to reduce inflammation across various neurological disorders [46]. A previous study investigated the beneficial effects of resveratrol in a SAH model, showing inhibition of the translocation of NF-
B, which was associated with reduced pro-inflammatory cytokines expression, reduced expression of MMP-9 and increased expression of junctional proteins [47]. All of these effects led to reduced mortality, improved neurological scores, reduced BBB disruption and consequent edema [47]. A later detailed experimental SAH study showed a significant reduction in acute inflammation upon resveratrol administration after SAH [46]. Resveratrol administration inhibited the expression of HMGB1, TLR-4, MyD88, NF-
B, which was associated with inhibition of microglial activation, reduced neuronal apoptosis, brain edema, BBB disruption and improved neurological scores [46]. Furthermore, resveratrol has been suggested to diminish adenosine diphosphate-induced platelet aggregation [31]. Consequently, inhibition of the platelet accumulation at the site of the lesion may be an additional mechanism, whereby resveratrol provides neuroprotection. Resveratrol effectively reduces the release of the pro-inflammatory cytokines [46][47]. Polyphenols are known to diminish the translocation of NF-κB into the nucleus, potentially inhibiting the expression of pro-inflammatory genes [46]. Very interestingly, a study investigating the role of curcumin in a SAH model has shown that curcumin alleviates inflammation by inhibiting the TLR-4/MyD88/NF-
B inflammatory axis and promotes the polarization of microglia towards a neuroprotective phenotype—the M2 (alternatively activated) microglia [44]. Further clinical investigations are required to determine the therapeutic effectiveness of these compounds.
4.5. Melatonin
Melatonin, a pleiotropic hormone produced by the pineal and extrapineal tissues regulates the circadian rhythms. However, this tryptophan derivative has important anti-inflammatory and immune modulatory properties [48][49][50]. In the SAH-induced rats by prechiasmatic cisternal injection of the autologous blood, treatment with melatonin has been shown to significantly reduce the expression of HMGB1, TLR4 and downstream proteins like NF-κB, MyD88, relevant pro-inflammatory cytokines such as IL-1β, TNF-α, IL-6, and inducible nitric oxide synthase. Importantly, the administration of melatonin after SAH significantly improved the memory and spatial learning as assessed by a water maze along with reduced neuronal apoptosis. This is consistent with the notion that melatonin displays neuro-protection not only via the anti-oxidant pathway, but also via anti-inflammatory signaling [51]. Clinical investigations aimed at melatonin-led improvements in post-SAH complications and clinical outcome stemming from reduced inflammation after SAH are required.
4.6. Monoclonal Antibodies
The activation of the MyD88 pathway enhances the expression of the pro-inflammatory cytokines. Direct cytokine inhibitors like monoclonal antibodies can be used for the significant reduction of SAH-associated inflammation. It was observed that the monoclonal antibody mediated targeting of TNF-α could be useful therapeutic target for SAH. Infliximab and canakinumab target the TNF-α and IL-1β, respectively [31]. The disadvantage of these antibodies are that they are mostly incapable of moving across the BBB and it requires extra measures to access the cerebral region. Nevertheless, in ischemic stroke, it was reported that infliximab can cross the BBB in the diseased condition. This is also true in case of SAH as the BBB is also disrupted and amenable to the therapeutic potential of these antibodies [8][31][52]. However, preliminary data suggests that the ICU stay of many SAH patients is usually prolonged because patients are often intubated and immobile, which greatly increases the chances of infection in the patients. Owing to the use of immunosuppressants, such patients are always prone to infections [31]. Therefore, cautious investigations regarding the clinical use of these immunosuppressing monoclonal antibodies are required without increasing the frequency of infections.
4.7. Pentoxifylline
Pentoxifylline is known as a nonselective phosphodiesterase inhibitor. Several studies have shown that pentoxifylline has anti-inflammatory and neuroprotective properties in a number of brain injury animal models. Pentoxifylline has been reported to inhibit the TLR4 and downstream signaling molecules such as MyD88 and NF-κB. Moreover, it possesses the potential to significantly reduce the pro-inflammatory cytokines, and reduce neural cell death and BBB permeability, highlighting another important clinically used drug that needs to be investigated in clinical trials in SAH patients [53].
4.8. Astaxanthin
Neuroinflammation is a hallmark of SAH and other cerebral injuries. Astaxanthin is a dietary carotenoid that possesses anti-inflammatory properties. Post-SAH treatment with astaxanthin can considerably down-regulate the MyD88 and NF-κB activity. Furthermore, inhibition of TLRs can improve cerebral edema, blood–brain barrier disruption, neurological dysfunction, and neuronal degeneration. As astaxanthin has anti-inflammatory potential and it retains neuro-protective potential against SAH, further investigations in clinical settings are needed [54].
4.9. Fluoxetine
Fluoxetine is a well-known antidepressant drug belonging to the selective serotonin reuptake inhibitors and its neuroprotective effects have been described in various neurological disorders [55]. Interestingly, fluoxetine administration in an endovascular perforation model of SAH has shown anti-inflammatory effects through the down regulation of TLR-4, MyD88, NF-κB, pro-inflammatory cytokines expression, reduced activation of microglia and infiltration of neutrophils, and improved neurobehavioral outcomes due to reduction in neuronal loss, brain edema and protection of junctional proteins [55]. Interestingly, fluoxetine also offers neuroprotection during early brain injury after SAH due to the inhibition of inflammasome formation and consequent necrotic death [55].
References
- Macdonald, R.L. Delayed neurological deterioration after subarachnoid haemorrhage. Nat. Rev. Neurol. 2014, 10, 44–58.
- Grasso, G.; Alafaci, C.; Macdonald, R.L. Management of aneurysmal subarachnoid hemorrhage: State of the art and future perspectives. Surg. Neurol. Int. 2017, 8, 11.
- Korja, M.; Kaprio, J. Controversies in epidemiology of intracranial aneurysms and SAH. Nat. Rev. Neurol. 2015, 12, 50–55.
- Gągało, I.; Rusiecka, I.; Kocic, I. Tyrosine Kinase Inhibitor as a new Therapy for Ischemic Stroke and other Neurologic Diseases: Is there any Hope for a Better Outcome? Curr. Neuropharmacol. 2015, 13, 836–844.
- Etminan, N.; Rinkel, G.J. Unruptured intracranial aneurysms: Development, rupture and preventive management. Nat. Rev. Neurol. 2016, 12, 699–713.
- Macdonald, R.L.; Diringer, M.N.; Citerio, G. Understanding the disease: Aneurysmal subarachnoid hemorrhage. Intensiv. Care Med. 2014, 40, 1940–1943.
- Chaudhry, S.R.; Frede, S.; Seifert, G.; Kinfe, T.M.; Niemelä, M.; Lamprecht, A.; Muhammad, S. Temporal profile of serum mitochondrial DNA (mtDNA) in patients with aneurysmal subarachnoid hemorrhage (aSAH). Mitochondrion 2019, 47, 218–226.
- Chaudhry, S.R.; Güresir, A.; Stoffel-Wagner, B.; Fimmers, R.; Kinfe, T.M.; Dietrich, D.; Lamprecht, A.; Vatter, H.; Güresir, E.; Muhammad, S. Systemic High-Mobility Group Box-1: A Novel Predictive Biomarker for Cerebral Vasospasm in Aneurysmal Subarachnoid Hemorrhage*. Crit. Care Med. 2018, 46, e1023–e1028.
- Chaudhry, S.R.; Hafez, A.; Jahromi, B.R.; Kinfe, T.M.; Lamprecht, A.; Niemelä, M.; Muhammad, S. Role of Damage Associated Molecular Pattern Molecules (DAMPs) in Aneurysmal Subarachnoid Hemorrhage (aSAH). Int. J. Mol. Sci. 2018, 19, 2035.
- Chaudhry, S.; Lehecka, M.; Niemelä, M.; Muhammad, S. Sterile Inflammation, Potential Target in Aneurysmal Subarachnoid Hemorrhage. World Neurosurg. 2019, 123, 159–160.
- Muhammad, S.; Chaudhry, S.R.; Kahlert, U.D.; Lehecka, M.; Korja, M.; Niemelä, M.; Hänggi, D. Targeting High Mobility Group Box 1 in Subarachnoid Hemorrhage: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 2709.
- Cahill, J.; Calvert, J.W.; Zhang, J.H. Mechanisms of early brain injury after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2006, 26, 1341–1353.
- Cahill, J.; Zhang, J.H. Subarachnoid Hemorrhage: Is It Time for a New Direction? Stroke 2008, 40 (Suppl. 1), S86–S87.
- Etminan, N.; Macdonald, R.L. Medical Complications After Aneurysmal Subarachnoid Hemorrhage: An Emerging Contributor to Poor Outcome. World Neurosurg. 2015, 83, 303–304.
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112.
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511.
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801.
- Zindel, J.; Kubes, P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 493–518.
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like receptors activation, signaling, and targeting: An overview. Bull. Natl. Res. Cent. 2019, 43, 1–12.
- Denning, N.-L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMPs and NETs in Sepsis. Front. Immunol. 2019, 10, 2536.
- Rahimifard, M.; Maqbool, F.; Moeini-Nodeh, S.; Niaz, K.; Abdollahi, M.; Braidy, N.; Nabavi, S.M. Targeting the TLR4 signaling pathway by polyphenols: A novel therapeutic strategy for neuroinflammation. Ageing Res. Rev. 2017, 36, 11–19.
- Muhammad, T.; Ikram, M.; Ullah, R.; Rehman, S.U.; Kim, M.O.J.N. Hesperetin, a citrus flavonoid, attenuates LPS-induced neuroinflammation, apoptosis and memory impairments by modulating TLR4/NF-κB signaling. Nutrients 2019, 11, 648.
- Janssens, S.; Beyaert, R. A universal role for MyD88 in TLR/IL-1R-mediated signaling. Trends Biochem. Sci. 2002, 27, 474–482.
- Gewies, A.; Ruland, J.; Kotlyarov, A.; Gaestel, M.; Procaccia, S.; Seger, R.; Yasuda, S.; Sugiura, H.; Yamagata, K.; Le, N.-T.; et al. MyD88, Myeloid Differentiation Primary Response Gene 88. In Encyclopedia of Signaling Molecules; Choi, S., Ed.; Springer: New York, NY, USA, 2012; pp. 1149–1159.
- Cheng, X.; Yang, Y.-L.; Yang, H.; Wang, Y.-H.; Du, G.-H. Kaempferol alleviates LPS-induced neuroinflammation and BBB dysfunction in mice via inhibiting HMGB1 release and down-regulating TLR4/MyD88 pathway. Int. Immunopharmacol. 2018, 56, 29–35.
- Vaure, C.; Liu, Y. A Comparative Review of Toll-Like Receptor 4 Expression and Functionality in Different Animal Species. Front. Immunol. 2014, 5, 316.
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151.
- Hanafy, K.A. The role of microglia and the TLR4 pathway in neuronal apoptosis and vasospasm after subarachnoid hemorrhage. J. Neuroinflamm. 2013, 10, 1–83.
- Wang, Q.; Luo, Q.; Zhao, Y.-H.; Chen, X. Toll-like receptor-4 pathway as a possible molecular mechanism for brain injuries after subarachnoid hemorrhage. Int. J. Neurosci. 2020, 130, 953–964.
- Kwon, M.S.; Woo, S.K.; Kurland, D.B.; Yoon, S.H.; Palmer, A.F.; Banerjee, U.; Iqbal, S.; Ivanova, S.; Gerzanich, V.; Simard, J.M. Methemoglobin Is an Endogenous Toll-Like Receptor 4 Ligand—Relevance to Subarachnoid Hemorrhage. Int. J. Mol. Sci. 2015, 16, 5028–5046.
- Karimy, J.K.; Reeves, B.C.; Kahle, K.T. Targeting TLR4-dependent inflammation in post-hemorrhagic brain injury. Expert Opin. Ther. Targets 2020, 24, 525–533.
- Sokół, B.; Wąsik, N.; Jankowski, R.; Hołysz, M.; Więckowska, B.; Jagodziński, P. Soluble Toll-Like Receptors 2 and 4 in Cerebrospinal Fluid of Patients with Acute Hydrocephalus following Aneurysmal Subarachnoid Haemorrhage. PLoS ONE 2016, 11, e0156171.
- Karimy, J.K.; Zhang, J.; Kurland, D.B.; Theriault, B.C.; Duran, D.; Stokum, J.A.; Furey, C.G.; Zhou, X.; Mansuri, M.S.; Montejo, J.; et al. Inflammation-dependent cerebrospinal fluid hypersecretion by the choroid plexus epithelium in posthemorrhagic hydrocephalus. Nat. Med. 2017, 23, 997–1003.
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145.
- Gay, N.J.; Symmons, M.F.; Gangloff, M.; Bryant, C.E. Assembly and localization of Toll-like receptor signalling complexes. Nat. Rev. Immunol. 2014, 14, 546–558.
- Fang, H.; Wang, P.-F.; Zhou, Y.; Wang, Y.-C.; Yang, Q.-W. Toll-like receptor 4 signaling in intracerebral hemorrhage-induced inflammation and injury. J. Neuroinflamm. 2013, 10, 27.
- Maddahi, A.; Ansar, S.; Chen, Q.; Edvinsson, L. Metabolism, Blockade of the MEK/ERK Pathway with a Raf Inhibitor Prevents Activation of Pro-Inflammatory Mediators in Cerebral Arteries and Reduction in Cerebral Blood Flow after Subarachnoid Hemorrhage in a Rat Model. Br. J. Pharmacol. 2010, 31, 144–154.
- Suzuki, H.; Hasegawa, Y.; Chen, W.; Kanamaru, K.; Zhang, J.H. Recombinant osteopontin in cerebral vasospasm after subarachnoid hemorrhage. Ann. Neurol. 2010, 68, 650–660.
- Zhu, Y.; Wu, M.; Wu, C.-Y.; Xia, G.-Q. Role of progesterone in TLR4-MyD88-dependent signaling pathway in pre-eclampsia. Acta Acad. Med. Wuhan 2013, 33, 730–734.
- Wang, Z.; Zuo, G.; Shi, X.-Y.; Zhang, J.; Fang, Q.; Chen, G. Progesterone Administration Modulates Cortical TLR4/NF-κB Signaling Pathway after Subarachnoid Hemorrhage in Male Rats. Mediat. Inflamm. 2011, 2011, 1–9.
- Yan, H.; Zhang, D.; Wei, Y.; Ni, H.; Liang, W.; Zhang, H.; Hao, S.; Jin, W.; Li, K.; Hang, C.-H. Inhibition of myeloid differentiation primary response protein 88 provides neuroprotection in early brain injury following experimental subarachnoid hemorrhage. Sci. Rep. 2017, 7, 15797.
- Fernández, G.; Moraga, A.; Cuartero, M.I.; García-Culebras, A.; Peña-Martínez, C.; Pradillo, J.M.; Hernández-Jiménez, M.; Sacristán, S.; Ayuso, M.I.; Gonzalo-Gobernado, R.J.M.T. TLR4-binding DNA aptamers show a protective effect against acute stroke in animal models. Mol. Ther. 2018, 26, 2047–2059.
- Jakus, P.B.; Kalman, N.; Antus, C.; Radnai, B.; Tucsek, Z.; Gallyas, F., Jr.; Sumegi, B.; Veres, B.J. TRAF6 is functional in inhibition of TLR4-mediated NF-κB activation by resveratrol. J. Nutr. Chem. 2013, 24, 819–823.
- Gao, Y.; Zhuang, Z.; Lu, Y.; Tao, T.; Zhou, Y.; Liu, G.; Wang, H.; Zhang, D.; Wu, L.; Dai, H.; et al. Curcumin Mitigates Neuro-Inflammation by Modulating Microglia Polarization Through Inhibiting TLR4 Axis Signaling Pathway Following Experimental Subarachnoid Hemorrhage. Front. Neurosci. 2019, 13, 1223.
- Wu, L.Y.; Ye, Z.N.; Zhuang, Z.; Gao, Y.; Tang, C.; Zhou, C.H.; Wang, C.X.; Zhang, X.S.; Xie, G.B.; Liu, J.P.; et al. Biochanin A Reduces Inflammatory Injury and Neuronal Apoptosis following Subarachnoid Hemorrhage via Suppression of the TLRs/TIRAP/MyD88/NF-kappaB Pathway. Behav. Neurol. 2018, 2018, 1960106.
- Zhang, X.-S.; Li, W.; Wu, Q.; Wu, L.-Y.; Ye, Z.-N.; Liu, J.-P.; Zhuang, Z.; Zhou, M.-L.; Zhang, X.; Hang, C.-H. Resveratrol Attenuates Acute Inflammatory Injury in Experimental Subarachnoid Hemorrhage in Rats via Inhibition of TLR4 Pathway. Int. J. Mol. Sci. 2016, 17, 1331.
- Shao, A.W.; Wu, H.J.; Chen, S.; Ammar, A.B.; Zhang, J.M.; Hong, Y. Resveratrol attenuates early brain injury after subarachnoid hemorrhage through inhibition of NF-κB-dependent inflammatory/MMP-9 pathway. CNS Neurosci. Ther. 2014, 20, 182–185.
- Hardeland, R. Melatonin and inflammation-Story of a double-edged blade. J. Pineal Res. 2018, 65, e12525.
- Tarocco, A.; Caroccia, N.; Morciano, G.; Wieckowski, M.R.; Ancora, G.; Garani, G.; Pinton, P. Melatonin as a master regulator of cell death and inflammation: Molecular mechanisms and clinical implications for newborn care. Cell Death Dis. 2019, 10, 1–12.
- Posa, L.; de Gregorio, D.; Gobbi, G.; Comai, S. Targeting Melatonin MT2 Receptors: A Novel Pharmacological Avenue for Inflammatory and Neuropathic Pain. Curr. Med. Chem. 2018, 25, 3866–3882.
- Wang, Z.; Wu, L.; You, W.; Ji, C.; Chen, G. Melatonin alleviates secondary brain damage and neurobehavioral dysfunction after experimental subarachnoid hemorrhage: Possible involvement of TLR4-mediated inflammatory pathway. J. Pineal Res. 2013, 55, 399–408.
- Tweedie, D.; Sambamurti, K.; Greig, N.H. TNF-α Inhibition as a Treatment Strategy for Neurodegenerative Disorders: New Drug Candidates and Targets. Curr. Alzheimer Res. 2007, 4, 378–385.
- Xia, D.Y.; Zhang, H.S.; Wu, L.Y.; Zhang, X.S.; Zhou, M.L.; Hang, C.H. Pentoxifylline Alleviates Early Brain Injury After Experimental Subarachnoid Hemorrhage in Rats: Possibly via Inhibiting TLR 4/NF-κB Signaling Pathway. Neurochem. Res. 2017, 42, 963–974.
- Zhang, X.; Lu, Y.; Wu, Q.; Dai, H.; Li, W.; Lv, S.; Zhou, X.; Zhang, X.; Hang, C.; Wang, J. Astaxanthin mitigates subarachnoid hemorrhage injury primarily by increasing sirtuin 1 and inhibiting the Toll-like receptor 4 signaling pathway. FASEB J. 2018, 33, 722–737.
- Liu, F.Y.; Cai, J.; Wang, C.; Ruan, W.; Guan, G.P.; Pan, H.Z.; Li, J.R.; Qian, C.; Chen, J.S.; Wang, L.; et al. Fluoxetine attenuates neuroinflammation in early brain injury after subarachnoid hemorrhage: A possible role for the regulation of TLR4/MyD88/NF-kappaB signaling pathway. J. Neuroinflamm. 2018, 15, 347.