Post-translational modifications (PTMs) add a layer of complexity to the proteome through the addition of biochemical moieties to specific residues of proteins, altering their structure, function and/or localization. Mass spectrometry (MS)-based techniques are at the forefront of PTM analysis due to their ability to detect large numbers of modified proteins with a high level of sensitivity and specificity.
- post-translational modifications
- blood cancer
- phosphorylation
- ubiquitination
- sumoylation
- acetylation
- multiple myeloma
- leukemia
- lymphoma
- myeloproliferative neoplasms
1. Introduction
The significance of proteome-focused studies in characterizing cellular phenotypes and disease mechanisms, in addition to discovering novel biomarkers and therapeutic targets, has been recognized in recent decades [1]. Proteomics involves the characterization and mapping of the proteome, a highly complex task due to temporal and spatial fluctuations in protein expression depending on the cellular environment. The post-translational modification (PTM) of proteins, a biochemical process that modifies the structure and function of a given protein, further enhances the complexity of the proteome [2]. Over 200 PTMs have been identified, many of which play vital roles in the regulation of various cellular processes such as cell growth, metabolism, differentiation, and apoptosis (
). Dysregulated PTMs alter the normal functioning of these processes which can contribute to the development and/or progression of disease, thus illustrating the need to identify and define primary PTM events associated with specific diseases such as blood cancers [3].
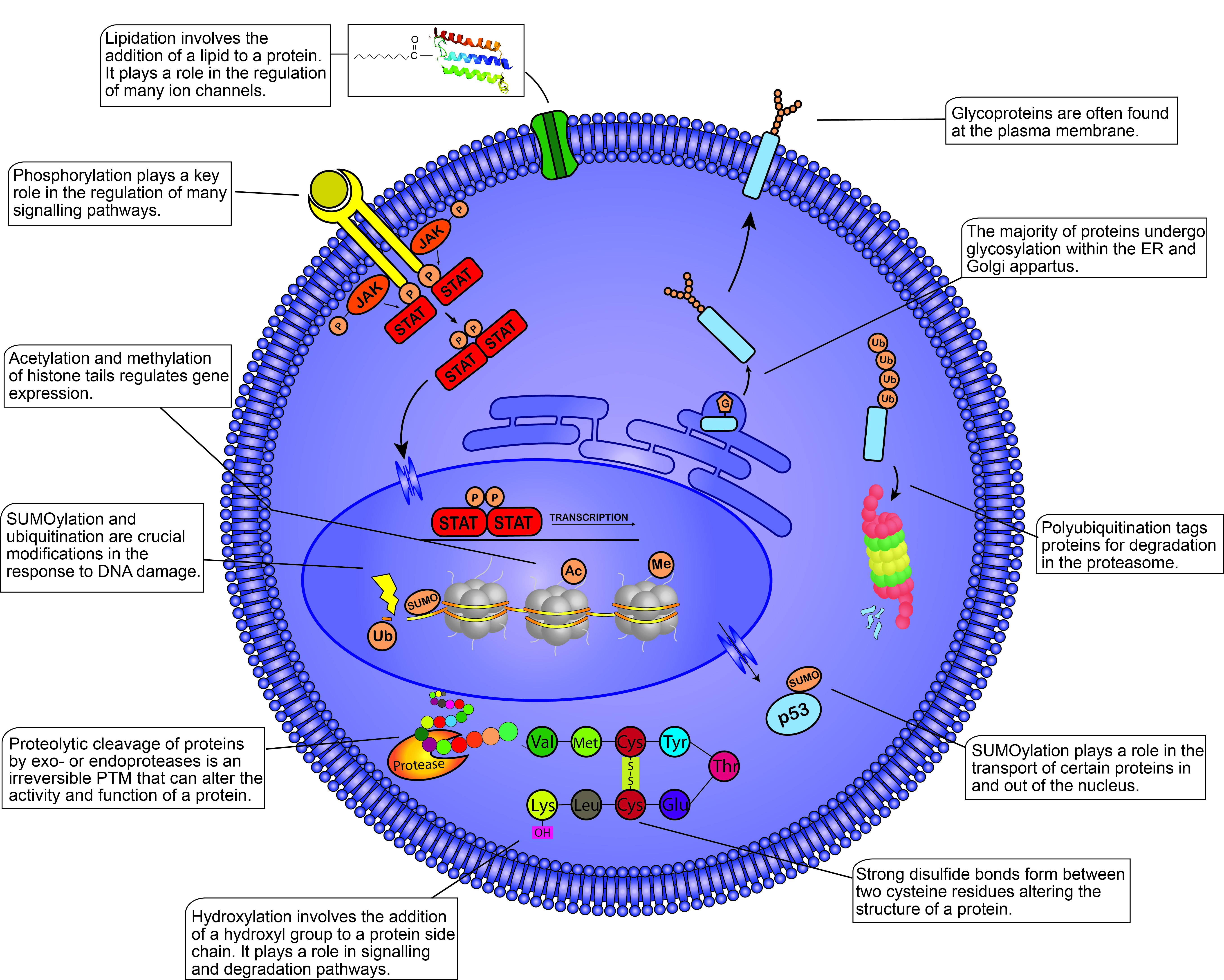
Post-translational modifications (PTMs) within the mammalian cell. This figure illustrates some of the most well-known PTMs and their functions within the cell. PTMs are found throughout the cell from the plasma membrane to the nucleosomes present within the nucleus. PTMs play crucial roles in almost all cellular processes including the cell cycle, degradation, apoptosis, cell signaling, transcription, etc. Different proteins modified by the same PTM will not always yield the same response, demonstrating the diverse functions of PTMs within the cell. JAK, Janus kinase; STAT, signal transducer and activator of transcription; P, phosphate group; G, glycosyl group; Ub, ubiquitin; SUMO, small ubiquitin-like modifier; Ac, acetyl group; Me, methyl group; Val, valine; Met, methionine; Cys, cysteine; Tyr, tyrosine; Glu, glutamic acid; Lys, lysine.
Hematological malignancies arise from blood or bone marrow cells that acquire genetic abnormalities resulting in uncontrolled proliferation, resisting cell death, and evading the immune response [4]. The incidence of hematological malignancies, which comprises a broad range of cancers including leukemias, lymphomas, multiple myeloma, myeloproliferative neoplasms (MPNs) and myelodysplastic syndromes (MDS), continues to rise, although advances in treatment have led to an increase in the five-year survival rate of many of these cancers [5]. In this review, we provide a comprehensive overview of current analytical techniques employed in the analysis of post-translational modifications and emphasize the contribution of these techniques to our current understanding of disease mechanisms associated with blood cancers. We highlight the central role of PTMs in the development of blood cancer therapeutics and consider the promising prospect of future improvements in the sensitivity and specificity of PTM-focused techniques to expedite our understanding of hematological malignancies and offer novel biological markers and therapeutic targets to augment the current arsenal of therapeutics available for the treatment of blood cancers.
2. Analytical Techniques in Post-Translational Modification Analysis
The most powerful analytical technique applied in proteomic studies is mass spectrometry (MS), which facilitates large-scale, highly specific, quantitative profiling of proteins and post-translationally modified proteins. Other techniques often used to study the proteome include flow cytometry, protein microarrays, Western blotting, and enzyme-linked immunosorbent assays (ELISA) (
) [1]. Additional experimental steps are often required to optimize proteomic techniques for PTM analysis due to the substoichiometric, transient and labile nature of PTMs which hinders the maintenance and detection of modifications during analysis [2]. To overcome the issue of low stoichiometry, enrichment of the PTM of interest is often performed prior to MS analysis. Modification-specific enrichment techniques such as ion exchange chromatography, immobilized metal ion affinity chromatography and immunoaffinity chromatography, separate modified proteins/peptides from their unmodified counterparts, reduce the complexity of the sample and increase the efficiency and reliability of the analysis (
) [3].
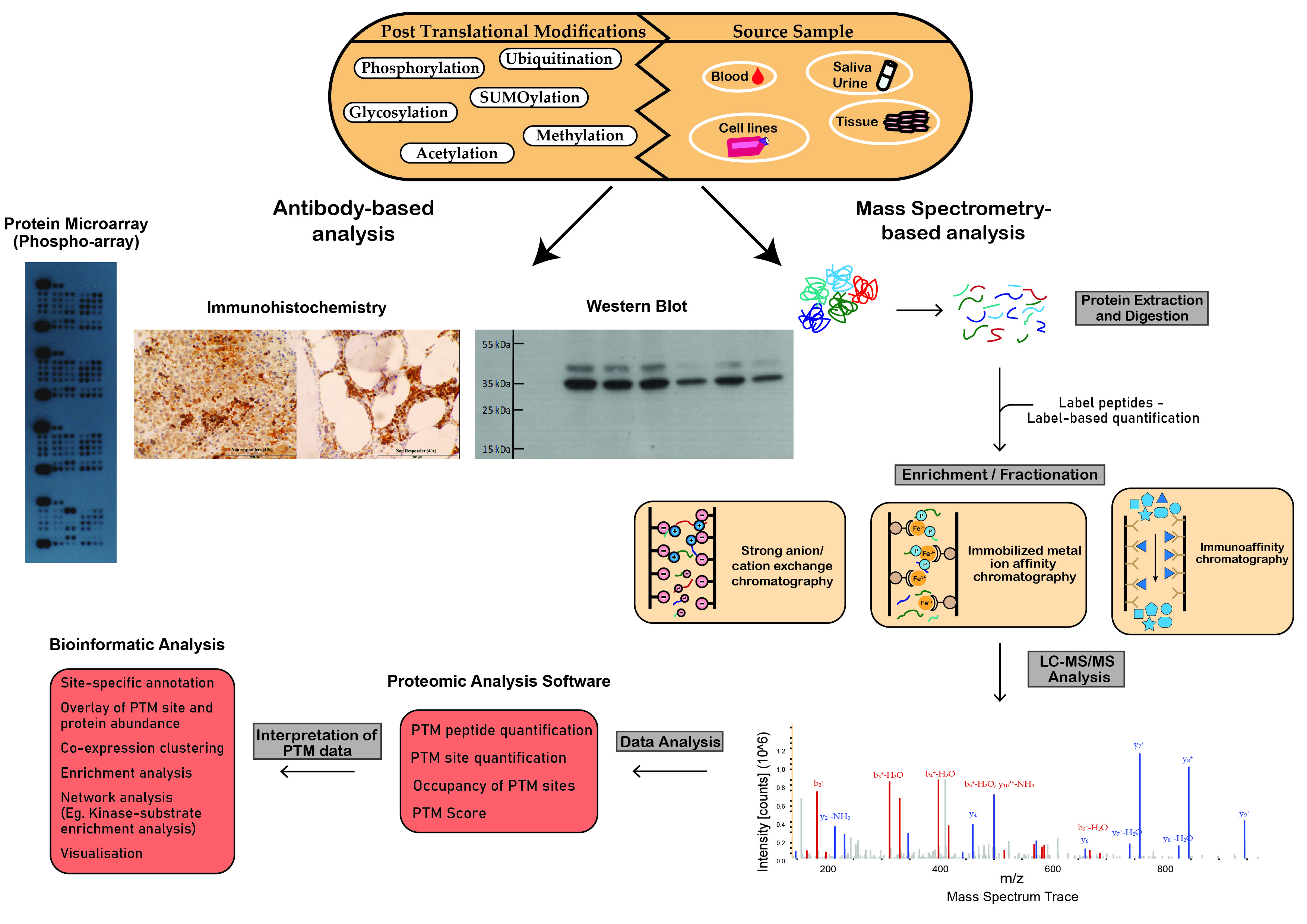
Schematic workflow illustrating analytical techniques used in the analysis of post-translational modifications. Proteomic techniques used for PTM analysis can be divided into antibody-based analysis or mass spectrometry (MS)-based analysis. Protein microarrays, immunohistochemistry and Western blot analysis demonstrate widespread popularity as efficient tools for antibody-based PTM analysis. MS-based analysis is a powerful technique in the study of PTMs. This figure outlines the steps involved in the preparation of crude samples for MS and the transformation of MS data into interpretable results.
Highly sensitive mass spectrometers with a high resolution, such as the Fourier transform (FT)-based mass spectrometers, are required for efficient PTM analysis. The “bottom-up” proteomics approach, typically used in PTM analysis, involves the cleavage of proteins in a biological sample by a protease, usually trypsin, prior to MS [6]. Sample preparation techniques, such as filter-aided sample preparation (FASP), separate peptides from undigested material in cell lysates [7]. Most MS-based proteomic protocols couple liquid chromatography with tandem MS (LC–MS/MS) to separate peptides before identification and quantitation by MS/MS. Following separation, the LC eluent is transferred to the ion source of the mass spectrometer where peptides are ionized, often via protonation [8]. High ionization efficiency enhances the sensitivity of the analysis; however, the efficiency of this step can be affected by the chemical nature of peptides being studied [9]. The dominant ionization technologies are matrix-assisted laser desorption/ionization (MALDI), electro-spray ionization (ESI) and atmospheric pressure chemical ionization (APCI) [10]. Following ionization, the peptide precursor ions are fragmented resulting in product ions that are subsequently characterized based on their mass-to-charge (m/z) ratios and relative abundance to produce MS/MS spectra (
Figure 2) [9]. Popular fragmentation techniques include collision-induced dissociation (CID) and high-energy collision dissociation (HCD). However, limitations, such as the loss of labile PTMs, mean alternative fragmentation methods, including electron capture/transfer dissociation (ECD/ETD), have been developed and incorporated into MS-based PTM analyses [11]. During CID and HCD, the collision of molecular ions results in cleavage of the amide bond in peptides whereas ETD involves the transfer of an electron to multiply protonated peptides resulting in the cleavage of the peptide backbone [12]. The more recently developed fragmentation methods, EThcD, which combines ETD and HCD, and activated ion ETD (AI-ETD) demonstrate great progress in combatting the challenges presented by conventional fragmentation techniques in PTMomics, such as uncertain site localizations [13,14,15]. The MS/MS spectra obtained during LC–MS/MS are matched to theoretical MS/MS spectra available on databases for peptide and PTM identification. Bioinformatic analysis, using algorithms that predict the probability of a PTM being present such as Ascore, PTMScore, PhosphoRS and Mascot Delta Score, are used to determine the validity of modification sites detected during LC–MS/MS [16].
) [9]. Popular fragmentation techniques include collision-induced dissociation (CID) and high-energy collision dissociation (HCD). However, limitations, such as the loss of labile PTMs, mean alternative fragmentation methods, including electron capture/transfer dissociation (ECD/ETD), have been developed and incorporated into MS-based PTM analyses [11]. During CID and HCD, the collision of molecular ions results in cleavage of the amide bond in peptides whereas ETD involves the transfer of an electron to multiply protonated peptides resulting in the cleavage of the peptide backbone [12]. The more recently developed fragmentation methods, EThcD, which combines ETD and HCD, and activated ion ETD (AI-ETD) demonstrate great progress in combatting the challenges presented by conventional fragmentation techniques in PTMomics, such as uncertain site localizations [13][14][15]. The MS/MS spectra obtained during LC–MS/MS are matched to theoretical MS/MS spectra available on databases for peptide and PTM identification. Bioinformatic analysis, using algorithms that predict the probability of a PTM being present such as Ascore, PTMScore, PhosphoRS and Mascot Delta Score, are used to determine the validity of modification sites detected during LC–MS/MS [16].
MS-based quantitation of proteins is typically divided into label-based and label-free techniques. Labeling peptides using differential mass tags, such as tandem mass tag (TMT) or isobaric tag for relative and absolute quantification (iTRAQ), allows for the comparison and multiplexing of multiple samples in a single MS run [17]. These isobaric labeling technologies involve the labelling of target proteins/peptides in different biological samples with tags made up of a reactive group, a mass normalization group, and a reporter group that have an identical overall mass but differ in the mass of the reporter group and mass normalization group. The reactive group facilitates the mass labeling of peptides. Fragmentation during tandem MS cleaves the reporter group, revealing reporter ions of variable molecular weights corresponding to the peptides present in the individual biological samples [18]. The in vitro labeling technique, stable-isotope labeling of amino acids in cell culture (SILAC), involves the labeling of peptides by growing an organism in cultures containing light or heavy stable isotope-containing amino acids (
13
C,
15
N), typically lysine and arginine, which are subsequently metabolically incorporated into proteins. During MS analysis, source samples can be easily differentiated by the isotopic mass shifts between the different isotopically labeled samples [19]. Subsequently, this technique has been adapted to facilitate the labelling of specific PTMs. For example,
13
C-glucose and D
3
-acetate,
13
CD
3
-methionine; and γ-
18
O
4-labeled ATP are added to cell culture media to label acetylation, methylation, and phosphorylation, respectively [20,21,22]. Label-free quantitation (LFQ) is a commonly used method of quantitation which refers to the use of peak intensity analysis and spectral counting for quantitation [23]. The results of qualitative or quantitative studies can undergo downstream statistical analysis to draw conclusions from the data through statistical tests, such as t-tests or ANOVAs, and/or visualize the data by creating networks, plots, and graphs [24].
-labeled ATP are added to cell culture media to label acetylation, methylation, and phosphorylation, respectively [20][21][22]. Label-free quantitation (LFQ) is a commonly used method of quantitation which refers to the use of peak intensity analysis and spectral counting for quantitation [23]. The results of qualitative or quantitative studies can undergo downstream statistical analysis to draw conclusions from the data through statistical tests, such as t-tests or ANOVAs, and/or visualize the data by creating networks, plots, and graphs [24].
A substantial effort has been made in recent years to develop specific antibodies for PTM analysis. However, low binding affinity is common among available antibodies due to the minuteness of PTM motifs, similarities in the chemical structure of certain PTMs, poor antigenicity and other difficulties in antibody generation [3]. Pan-PTM-specific antibodies are often applied for immunoaffinity enrichment prior to LC–MS/MS as well as Western blotting, protein microarrays, immunohistochemistry, and flow cytometry. Antibodies against specific modification sites known to play an important biological role are also available for certain modifications including phosphorylation, methylation, and acetylation [25,26,27]. Other PTMs face difficulties in site-specific antibody generation due to the size or transience of the PTM (e.g., ubiquitination); or lack sufficient evidence of site occupancy to invest in antibody generation [28].
A substantial effort has been made in recent years to develop specific antibodies for PTM analysis. However, low binding affinity is common among available antibodies due to the minuteness of PTM motifs, similarities in the chemical structure of certain PTMs, poor antigenicity and other difficulties in antibody generation [3]. Pan-PTM-specific antibodies are often applied for immunoaffinity enrichment prior to LC–MS/MS as well as Western blotting, protein microarrays, immunohistochemistry, and flow cytometry. Antibodies against specific modification sites known to play an important biological role are also available for certain modifications including phosphorylation, methylation, and acetylation [25][26][27]. Other PTMs face difficulties in site-specific antibody generation due to the size or transience of the PTM (e.g., ubiquitination); or lack sufficient evidence of site occupancy to invest in antibody generation [28].
Flow cytometry (FCM) is a scientific technique used to measure the physical and biochemical characteristics of cells, including size, internal complexity, and cellular components such as DNA, RNA, and specific proteins on the cell surface or intracellularly. FCM has gained prominence in recent years and is now routinely used in basic research and clinical practice, particularly in the oncology and immunology settings. FCM is an extremely sensitive technique, capable of detecting an extremely small number of cancer cells within a dense cellular network. This technique also provides a quick turnaround time, with the ability to measure a significant number of parameters in thousands of cells in a matter of seconds. The detection of surface antigens by FCM can be used in real time to physically sort cells into specific populations, a process known as cell sorting or fluorescence-activated cell sorting (FACS). This is particularly useful for the analysis of hematological malignancies with immunophenotyping by FCM often used in the clinical diagnosis and subclassification of leukemias and lymphomas. Once the cells of interest are selected, qualitative information may be obtained by measuring fluorescence intensity for specific antigens, for example acute leukemia (CD45), chronic lymphocytic leukemia (CD19/CD5), and Non-Hodgkin’s lymphoma (CD20) [29,30]. FCM-based minimal residual disease (MRD) assays are powerful diagnostic and disease monitoring tools in multiple types of blood cancers. By detecting low-frequency malignant cells (ratio as low as 1:10000 compared to normal cells), this approach is critical in the clinical management of patients [31]. As MRD status appears to correlate with patient outcomes in some hematological malignancies, increasing the sensitivity and robustness of MRD assays analyzed by FCM is currently an active area of research. Ongoing improvements in the quality of PTM site-specific antibodies will strengthen FCM as a promising method for characterization of cellular activities and signal transduction pathways in hematological malignancies in research and clinical settings.
Flow cytometry (FCM) is a scientific technique used to measure the physical and biochemical characteristics of cells, including size, internal complexity, and cellular components such as DNA, RNA, and specific proteins on the cell surface or intracellularly. FCM has gained prominence in recent years and is now routinely used in basic research and clinical practice, particularly in the oncology and immunology settings. FCM is an extremely sensitive technique, capable of detecting an extremely small number of cancer cells within a dense cellular network. This technique also provides a quick turnaround time, with the ability to measure a significant number of parameters in thousands of cells in a matter of seconds. The detection of surface antigens by FCM can be used in real time to physically sort cells into specific populations, a process known as cell sorting or fluorescence-activated cell sorting (FACS). This is particularly useful for the analysis of hematological malignancies with immunophenotyping by FCM often used in the clinical diagnosis and subclassification of leukemias and lymphomas. Once the cells of interest are selected, qualitative information may be obtained by measuring fluorescence intensity for specific antigens, for example acute leukemia (CD45), chronic lymphocytic leukemia (CD19/CD5), and Non-Hodgkin’s lymphoma (CD20) [29][30]. FCM-based minimal residual disease (MRD) assays are powerful diagnostic and disease monitoring tools in multiple types of blood cancers. By detecting low-frequency malignant cells (ratio as low as 1:10000 compared to normal cells), this approach is critical in the clinical management of patients [31]. As MRD status appears to correlate with patient outcomes in some hematological malignancies, increasing the sensitivity and robustness of MRD assays analyzed by FCM is currently an active area of research. Ongoing improvements in the quality of PTM site-specific antibodies will strengthen FCM as a promising method for characterization of cellular activities and signal transduction pathways in hematological malignancies in research and clinical settings.
When designing a PTM-focused experiment, researchers must consider additional sample preparation steps that may be required to analyze modifications on proteins from specific cellular components, such as the cell surface, extracellular vesicles, and the nucleus. Histone proteins represent a well-known target for PTM-focused analyses as they are extensively modified by various PTMs to facilitate dynamic genomic organization and transcriptional regulation. PTMs found on histones include methylation, acetylation and ubiquitination on lysine residues, phosphorylation on serine, threonine, and tyrosine residues and/or citrullination on arginine residues [32]. Histone PTMs are routinely detected through site-specific antibodies, as demonstrated recently by Xu and co-workers [33] investigating histone H3 lysine 36 dimethylation (H3K36me2) levels in myeloma cells under normoxia or hypoxia conditions using Western blotting [33]. By detecting both the protein and site-specific modification abundance levels using precise antibodies, the degree of histone modification can be quantified. However, in the past, some problems with respect to anti-PTM histone antibody quality surfaced, facilitating the need to develop alternative platforms [34]. Mass spectrometry has proven to be an invaluable research platform in identifying and quantifying histone PTMs. Several histone enrichment methods are available prior to analysis, including chromatin affinity purification with MS (ChAP–MS), an approach that facilitates the isolation of targeted chromatin sections for the identification of histone PTMs [35]. Another isolation method involves the use of acidic or high-salt conditions to extract histones from cell lysates [32]. Due to the high ratio of the basic amino acids, arginine and lysine, in histones, tryptic digestion generates short peptides (2–4 amino acids) that are incompatible with LC–MS/MS analysis. Thus, chemical derivatization, often via propionylation, of lysine residues has been implemented to inhibit cleavage after lysine, resulting in peptides with lengths compatible with LC–MS/MS [36]. Detection of histone PTMs from MS/MS spectra is accomplished by detecting mass shifts corresponding to the PTM of interest, as described in the sections below, to confirm the presence of the modification. Determining the function of histone PTMs often involves investigating interacting partners, in particular the suite of associated enzymes responsible for the regulation of site-specific modifications. Crosslinking mass spectrometry (XL–MS) has emerged as a powerful approach to elucidate protein–protein complexes by mass spectrometry, with applications for structural mechanism discovery involving histone activity [37].
3. Application of PTM-Focused Techniques in Blood Cancer Research
Several reviews have been published focusing on specific PTMs and their involvement in carcinogenesis. However, literature surrounding the relationship between PTMs and hematological malignancies is limited [186,187,188]. Both the individual PTMs and associated modifying enzymes play significant roles as biomarkers and therapeutic targets in hematological malignancies [189]. Many signaling pathways regulated by PTMs have demonstrated aberrant activity in blood cancers, such as the Janus Kinase (JAK)/signal transducer and activator of transcription (STAT), phosphatidylinositol-3-kinase (PI3K)/AKT and B-cell receptor (BCR) signaling pathways (
Several reviews have been published focusing on specific PTMs and their involvement in carcinogenesis. However, literature surrounding the relationship between PTMs and hematological malignancies is limited [38][39][40]. Both the individual PTMs and associated modifying enzymes play significant roles as biomarkers and therapeutic targets in hematological malignancies [41]. Many signaling pathways regulated by PTMs have demonstrated aberrant activity in blood cancers, such as the Janus Kinase (JAK)/signal transducer and activator of transcription (STAT), phosphatidylinositol-3-kinase (PI3K)/AKT and B-cell receptor (BCR) signaling pathways (
Figure 4). Dysregulation of these pathways cause proliferation, migration, survival, and angiogenesis which promote and/or maintain the malignant phenotype of malignant blood cells. Several genetic abnormalities associated with hematological malignancies have also been found to affect protein modifier genes, such as the t(9;22) translocation which results in the oncogenic BCR-ABL fusion kinase (
3). Dysregulation of these pathways cause proliferation, migration, survival, and angiogenesis which promote and/or maintain the malignant phenotype of malignant blood cells. Several genetic abnormalities associated with hematological malignancies have also been found to affect protein modifier genes, such as the t(9;22) translocation which results in the oncogenic BCR-ABL fusion kinase (
Figure 4) [190]. Already, many FDA-approved therapies for hematological malignancies incorporate the modulation or inhibition of oncogenic protein modifications as part of their mode of action (
3) [42]. Already, many FDA-approved therapies for hematological malignancies incorporate the modulation or inhibition of oncogenic protein modifications as part of their mode of action (
,
Figure 4). Therefore, the application of efficient analytical techniques in laboratories globally for thorough PTM analysis may aid in the discovery of novel biomarkers and therapeutic targets in hematological malignancies. MS-based techniques are typically used for large-scale systematic analysis of PTM marks in blood cancers, generating extensive data. Bioinformatic analysis of this data can identify significant changes in the patterns of protein modification between disease states, such as malignant versus normal samples. Further research can then be conducted to determine the importance and implications of specific PTM changes in blood cancers. The technique of choice for validation and small-scale analyses is Western blot analysis using antibodies specific to the PTM being analyzed.
3). Therefore, the application of efficient analytical techniques in laboratories globally for thorough PTM analysis may aid in the discovery of novel biomarkers and therapeutic targets in hematological malignancies. MS-based techniques are typically used for large-scale systematic analysis of PTM marks in blood cancers, generating extensive data. Bioinformatic analysis of this data can identify significant changes in the patterns of protein modification between disease states, such as malignant versus normal samples. Further research can then be conducted to determine the importance and implications of specific PTM changes in blood cancers. The technique of choice for validation and small-scale analyses is Western blot analysis using antibodies specific to the PTM being analyzed.
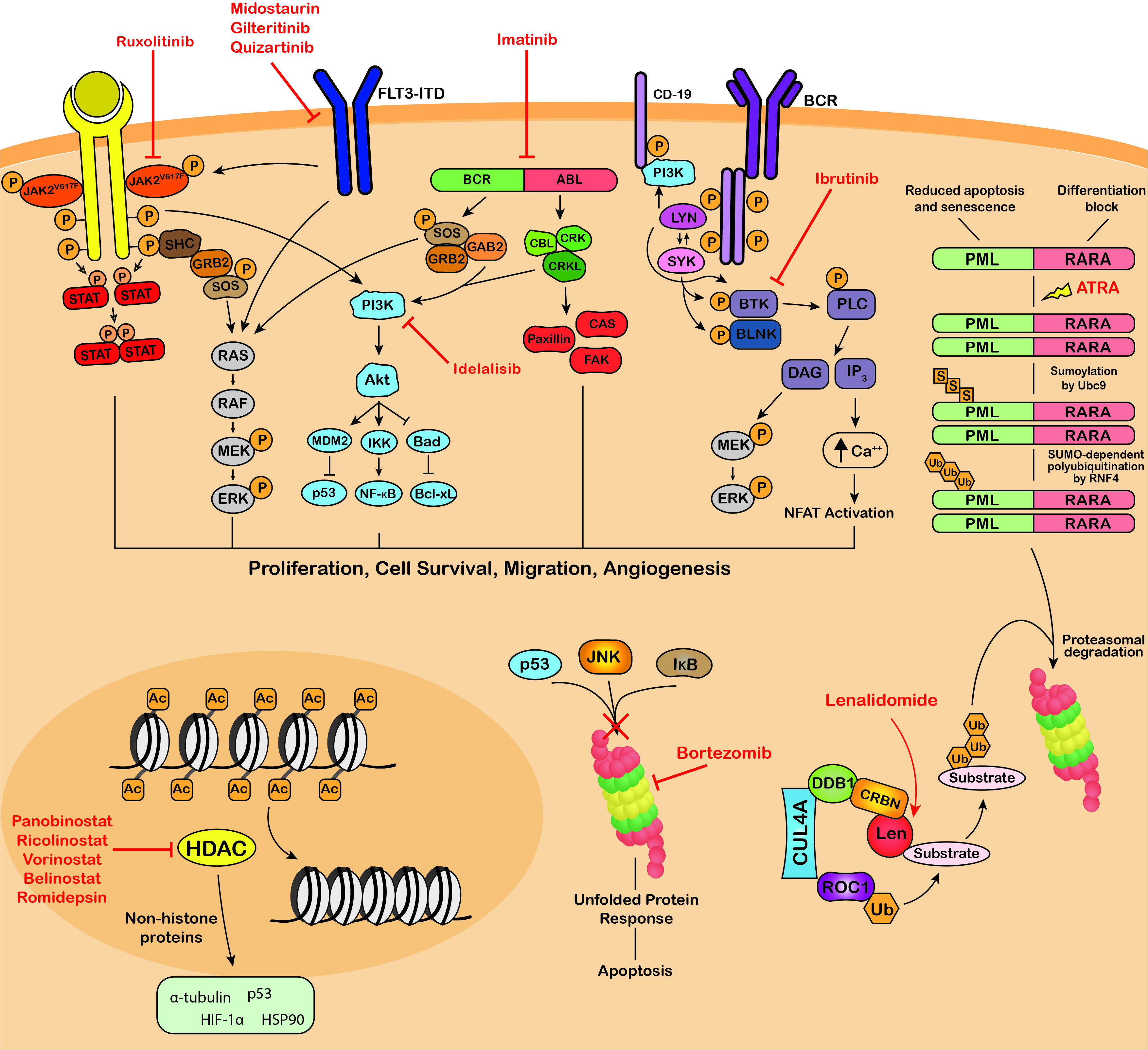
Targeting of protein modifications by clinically used blood cancer therapeutics. Ruxolitinib, midostaurin, gilteritinib, imatinib, dasatinib, nilotinib, bosutinib, ponatinib, ibrutinib and idelalisib are kinase inhibitors that inhibit the phosphorylation and activation of key proteins of oncogenic signaling pathways, promoting growth arrest and apoptosis in cancer cells. ATRA therapy for the treatment of APL induces SUMOylation-dependent polyubiquitination and proteasomal degradation of the fusion oncoprotein PML-RARA. The immunomodulatory drugs, lenalidomide, thalidomide and pomalidomide bind Cereblon, the substrate adaptor of the CRL4
E3 ubiquitin ligase, modulating its substrate specificity. Bortezomib, carfilzomib and ixazomib target the 26S proteasome, blocking the degradation of polyubiquitinated regulatory proteins and inducing the unfolded protein response resulting in apoptosis. Panobinostat, vorinostat, belinostat and romidepsin are HDAC inhibitors that block the deacetylation activity of HDACs and exert their anti-tumor activity through modulating the acetylation status of a variety of histone and non-histone proteins. JAK2
, Janus kinase 2 V617F; STAT, signal transducer and activator of transcription; SHC, Src homology and Collagen; GRB2, growth factor receptor-bound protein 2; SOS, Son of sevenless; RAF, RAF proto-oncogene serine/threonine-protein kinase; MEK, Mitogen-activated protein kinase kinase; ERK, extracellular signal-regulated kinase; FLT3-ITD, fms like tyrosine kinase 3 – internal tandem duplications; PI3K, phosphatidylinositol 3-kinase; MDM2, mouse double minute 2 homolog; IKK, IκB kinase; Bad, BCL2 associated agonist of cell death; NF-κB, Nuclear factor kappa B; Bcl-xL, B-cell lymphoma-extra large; GAB2, GRB2 associated binding protein 2; BCR-ABL, breakpoint cluster region-proto-oncogene tyrosine-protein kinase; CBL, E3 ubiquitin-protein ligase CBL; CRK, Proto-oncogene c-Crk; CRKL, Crk-like protein; CAS, Crk-associated substrate; FAK, focal adhesion kinase; BCR, B-cell receptor; Lyn, tyrosine-protein kinase Lyn; SYK, tyrosine-protein kinase SYK; BTK, Bruton’s tyrosine kinase; BLNK, B-cell linker protein; PLC, phospholipase C; DAG, diacyl glycerol; IP
, inositol 1,4,5-trisphosphate; NFAT, nuclear factor of activated T-cells; PML-RARA, promyelocytic leukemia/retinoic acid receptor alpha; ATRA, all-trans retinoic acid; Ubc9, SUMO-conjugating enzyme UBC9; RNF4, ring finger protein 4; CUL4A, cullin 4A; DDB1, DNA damage binding protein 1; CRBN, cereblon; ROC1, regulator of cullins-1; Len, lenalidomide; JNK, c-Jun N-terminal kinase (JNK); IκB, inhibitor of nuclear factor kappa B; HIF-1α, hypoxia-inducible factor 1-alpha; HSP90, heat shock protein 90; P, phosphorylation; S, sumoylation; Ub, ubiquitination; Ac, acetylation.
Current FDA-approved therapeutics for the treatment of various blood cancers that influence protein modifications.
| Type of Therapeutic | Drug | PTM Affected | Type of Blood Cancer | Mechanism of Action | References |
|---|---|---|---|---|---|
| Kinase Inhibitors (KIs) | Ruxolitinib (JAKAFI®) | Phosphorylation | Myelofibrosis Polycythemia Vera |
JAK2 inhibitor | [191,192][43][44] |
| Midostaurin (RYDAPT®) | Phosphorylation | FLT3-mutant AML, Advanced systemic mastocytosis (AdvSM) |
FLT3 inhibitor in AML. KIT inhibitor in AdvSM. |
[193,194][45][46] | |
| Gilteritinib (XOSPATA®) | Phosphorylation | FLT3-mutant Acute myeloid leukemia (AML) | FLT3, ALK inhibitor | [195][47] | |
| Imatinib (GLEEVEC®) | Phosphorylation | Ph+ Chronic myeloid leukemia (CML), Ph+ Acute lymphoblastic leukemia (ALL), Myelodysplastic/ myeloproliferative diseases (MDS/MPD), Aggressive systemic mastocytosis (ASM), Chronic eosinophilic leukemia (CEL) |
BCR-ABL inhibitor | [196][48] | |
| Dasatinib (SPRYCEL®) | Phosphorylation | Ph+ CML, Ph+ ALL | BCR-ABL, SRC inhibitor | [197][49] | |
| Nilotinib (TASIGNA®) | Phosphorylation | Ph+ CML | BCR-ABL inhibitor | [198][50] | |
| Bosutinib (BOSULIF®) | Phosphorylation | Ph+ CML | BCR-ABL and SRC inhibitor | [199][51] | |
| Ponatinib (ICLUSIG®) | Phosphorylation | CML, Ph+ ALL | BCR-ABL inhibitor | [200][52] | |
| Ibrutinib (IMBRUVICA®) | Phosphorylation | Mantle cell lymphoma (MCL), Chronic lymphocytic leukaemia (CLL) Small lymphocytic lymphoma (SLL) Waldenström’s macroglobulinemia (WM) Marginal zone lymphoma (MZL) |
BTK inhibitor | [201][53] | |
| Idelalisib (ZYDELIG®) | Phosphorylation | CLL, SLL Follicular lymphoma (FL) |
Phosphatidylinositol 3-kinase delta (PI3Kδ) inhibitor | [202][54] | |
| Proteasome Inhibitors (PIs) | Bortezomib (VELCADE®) | Ubiquitination | MCL, Multiple myeloma (MM) |
26S proteasome inhibitor | [203][55] |
| Carfilzomib (KYPROLIS®) | Ubiquitination | MM | 26S proteasome inhibitor | [204][56] | |
| Ixazomib (NINLARO®) | Ubiquitination | MM | 26S proteasome inhibitor | [205][57] | |
| Differentiation Therapy | All-trans retinoic acid (ATRA) (VESANOID®) and arsenic trioxide (TRISENOX®) | Sumoylation Ubiquitination |
Acute promyelocytic leukemia (APL) | Sumoylation-dependent degradation of the fusion oncoprotein PML-RARα. | [206][58] |
| Immunomodulatory Drugs (IMiDs) | Lenalidomide (REVLIMID®) | Ubiquitination | MM, MDS, MCL, FL, MZL | Modulation of CRL4CRBN E3 ubiquitin ligase activity. | [207][59] |
| Thalidomide (THALOMID®) | Ubiquitination | MM | Modulation of CRL4CRBN E3 ubiquitin ligase activity. | [208][60] | |
| Pomalidomide (POMALYST®) | Ubiquitination | MM | Modulation of CRL4CRBN E3 ubiquitin ligase activity. | [208][60] | |
| Histone Deacetylase Inhibitors (HDACi) | Panobinostat (FARYDAK®) | Acetylation | MM | Pan-HDAC inhibitor | [209][61] |
| Vorinostat (ZOLINZA®) | Acetylation | Cutaneous T cell lymphoma (CTCL) | Class I, II HDAC inhibitor | [210][62] | |
| Belinostat (BELEODAQ®) | Acetylation | Peripheral T cell lymphoma (PTCL) | Pan-HDAC inhibitor | [211][63] | |
| Romidepsin (ISTODAX®) | Acetylation | CTCL, PTCL | Class I HDAC inhibitor | [212][64] |
4. Conclusions
The adaptation and fine tuning of conventional proteomic techniques for use in the analysis of PTMs have seen significant progress in recent decades. The widespread availability of highly sensitive equipment capable of high-throughput analyses has led to breakthroughs in cancer research and the application of these techniques in blood cancer research is limitless. The analysis of PTMs, thus far, has opened avenues for translational research into the development of novel biomarker signatures and therapeutics to improve patient survival. Many studies have focused on phosphorylation and ubiquitination analysis. However, recently, the targeting of less common PTMs, such as SUMOylation, has been demonstrated as a promising approach for the treatment of hematological malignancies. Continuous improvements in the analytical power of mass spectrometry with simultaneous optimization of bioinformatic pipelines and enhanced sensitivity and specificity of PTM-specific antibodies will facilitate deeper analysis of obscure modifications that will indefinitely provide meaning to many molecular processes occurring in the cellular environment. Future applications of the proteomic techniques described in this review will enhance our overall understanding of hematological malignancies, leading to improvements in therapies and, thus, patient survival.
References
- Aslam, B.; Basit, M.; Nisar, M.A.; Khurshid, M.; Rasool, M.H. Proteomics: Technologies and Their Applications. J. Chromatogr. Sci. 2017, 55, 182–196.
- Yakubu, R.R.; Nieves, E.; Weiss, L.M. The Methods Employed in Mass Spectrometric Analysis of Posttranslational Modifications (PTMs) and Protein–Protein Interactions (PPIs). Adv. Exp. Med. Biol. 2019, 1140, 169–198.
- Zhao, Y.; Jensen, O.N. Modification-specific proteomics: Strategies for characterization of post-translational modifications using enrichment techniques. Proteomics 2009, 9, 4632–4641.
- Rahman, S.; Mansour, M.R. The role of noncoding mutations in blood cancers. Dis. Model. Mech. 2019, 12, dmm041988.
- Pulte, D.; Jansen, L.; Brenner, H. Changes in long term survival after diagnosis with common hematologic malignancies in the early 21st century. Blood Cancer J. 2020, 10, 1–8.
- Doll, S.; Gnad, F.; Mann, M. The Case for Proteomics and Phospho-Proteomics in Personalized Cancer Medicine. Proteom. Clin. Appl. 2019, 13, e1800113.
- Wiśniewski, J.R. Filter Aided Sample Preparation—A tutorial. Anal. Chim. Acta 2019, 1090, 23–30.
- Sinha, A.; Mann, M. A beginner’s guide to mass spectrometry–based proteomics. Biochemist 2020, 42, 64–69.
- Larsen, M.R.; Trelle, M.B.; Thingholm, T.E.; Jensen, O.N. Analysis of posttranslational modifications of proteins by tandem mass spectrometry. BioTechniques 2006, 40, 790–798.
- Siuzdak, G. An introduction to mass spectrometry ionization: An excerpt from The Expanding Role of Mass Spectrometry in Biotechnology, 2nd ed.; MCC Press: San Diego, 2005. J. Lab. Autom. 2004, 9, 50–63.
- Fort, K.L.; Cramer, C.N.; Voinov, V.G.; Vasil’Ev, Y.V.; Lopez, N.I.; Beckman, J.S.; Heck, A.J.R. Exploring ECD on a Benchtop Q Exactive Orbitrap Mass Spectrometer. J. Proteome Res. 2017, 17, 926–933.
- Penkert, M.; Hauser, A.; Harmel, R.; Fiedler, D.; Hackenberger, C.P.R.; Krause, E. Electron Transfer/Higher Energy Collisional Dissociation of Doubly Charged Peptide Ions: Identification of Labile Protein Phosphorylations. J. Am. Soc. Mass Spectrom. 2019, 30, 1578–1585.
- Frese, C.K.; Zhou, H.; Taus, T.; Altelaar, A.F.M.; Mechtler, K.; Heck, A.J.R.; Mohammed, S. Unambiguous Phosphosite Localization using Electron-Transfer/Higher-Energy Collision Dissociation (EThcD). J. Proteome Res. 2013, 12, 1520–1525.
- Riley, N.M.; Hebert, A.S.; Dürnberger, G.; Stanek, F.; Mechtler, K.; Westphall, M.S.; Coon, J.J. Phosphoproteomics with Activated Ion Electron Transfer Dissociation. Anal. Chem. 2017, 89, 6367–6376.
- Yu, Q.; Wang, B.; Chen, Z.; Urabe, G.; Glover, M.S.; Shi, X.; Guo, L.-W.; Kent, K.C.; Li, L. Electron-Transfer/Higher-Energy Collision Dissociation (EThcD)-Enabled Intact Glycopeptide/Glycoproteome Characterization. J. Am. Soc. Mass Spectrom. 2017, 28, 1751–1764.
- Chalkley, R.J.; Clauser, K.R. Modification Site Localization Scoring: Strategies and Performance. Mol. Cell. Proteom. 2012, 11, 3–14.
- Megger, D.A.; Pott, L.L.; Ahrens, M.; Padden, J.; Bracht, T.; Kuhlmann, K.; Eisenacher, M.; Meyer, H.E.; Sitek, B. Comparison of label-free and label-based strategies for proteome analysis of hepatoma cell lines. Biochim. Biophys. Acta (BBA)Proteins Proteom. 2014, 1844, 967–976.
- Anand, S.; Samuel, M.; Ang, C.-S.; Keerthikumar, S.; Mathivanan, S. Label-Based and Label-Free Strategies for Protein Quantitation. Adv. Struct. Saf. Stud. 2016, 1549, 31–43.
- Ong, S.-E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol. Cell. Proteom. 2002, 1, 376–386.
- Nemutlu, E.; Zhang, S.; Gupta, A.; Juranic, N.O.; Macura, S.I.; Terzic, A.; Jahangir, A.; Dzeja, P.P. Dynamic phosphometabolomic profiling of human tissues and transgenic models by18O-assisted31P NMR and mass spectrometry. Physiol. Genom. 2012, 44, 386–402.
- Liao, H.; Zang, Q.; Lv, Q.; Gao, Y.; Zhao, Z.; He, J.; Zhang, R.; Song, Y.; Chen, Y.; Abliz, Z. Development of methionine methylation profiling and relative quantification in human breast cancer cells based on metabolic stable isotope labeling. Analyst 2019, 144, 3988–3998.
- Lund, P.J.; Kori, Y.; Zhao, X.; Sidoli, S.; Yuan, Z.-F.; Garcia, B.A. Isotopic Labeling and Quantitative Proteomics of Acetylation on Histones and Beyond. Breast Cancer 2019, 1977, 43–70.
- Filiou, M.D.; Martins-De-Souza, D.; Guest, P.C.; Bahn, S.; Turck, C.W. To label or not to label: Applications of quantitative proteomics in neuroscience research. Proteomics 2012, 12, 736–747.
- Refsgaard, J.C.; Munk, S.; Jensen, L.J. Search Databases and Statistics: Pitfalls and Best Practices in Phosphoproteomics. Adv. Struct. Safety Stud. 2016, 1355, 323–339.
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837.
- Jiang, Y.-Y.; Jiang, Y.; Li, C.-Q.; Zhang, Y.; Dakle, P.; Kaur, H.; Deng, J.-W.; Lin, R.Y.-T.; Han, L.; Xie, J.-J.; et al. TP63, SOX2, and KLF5 Establish a Core Regulatory Circuitry That Controls Epigenetic and Transcription Patterns in Esophageal Squamous Cell Carcinoma Cell Lines. Gastroenterology 2020, 159, 1311–1327.e19.
- Kim, S.W.; Hasanuzzaman, M.; Cho, M.; Heo, Y.R.; Ryu, M.-J.; Ha, N.-Y.; Park, H.J.; Park, H.-Y.; Shin, J.-G. Casein Kinase 2 (CK2)-mediated Phosphorylation of Hsp90β as a Novel Mechanism of Rifampin-induced MDR1 Expression. J. Biol. Chem. 2015, 290, 17029–17040.
- Van Kruijsbergen, I.; Mulder, M.P.C.; Uckelmann, M.; Van Welsem, T.; De Widt, J.; Spanjaard, A.; Jacobs, H.; El Oualid, F.; Ovaa, H.; Van Leeuwen, F. Strategy for Development of Site-Specific Ubiquitin Antibodies. Front. Chem. 2020, 8, 111.
- Keeney, M.; Hedley, B.D.; Chin-Yee, I.H. Flow cytometry-Recognizing unusual populations in leukemia and lymphoma diagnosis. Int. J. Lab. Hematol. 2017, 39, 86–92.
- Heo, S.-K.; Noh, E.-K.; Ju, L.J.; Sung, J.Y.; Jeong, Y.K.; Cheon, J.; Koh, S.J.; Min, Y.J.; Choi, Y.; Jo, J.-C. CD45dimCD34+CD38−CD133+ cells have the potential as leukemic stem cells in acute myeloid leukemia. BMC Cancer 2020, 20, 1–10.
- Ngai, L.L.; Kelder, A.; Janssen, J.J.W.M.; Ossenkoppele, G.J.; Cloos, J. MRD Tailored Therapy in AML: What We Have Learned So Far. Front. Oncol. 2021, 10, 603636.
- Shechter, D.; Dormann, H.L.; Allis, C.D.; Hake, S.B. Extraction, purification and analysis of histones. Nat. Protoc. 2007, 2, 1445–1457.
- Xu, Y.; Guo, J.; Liu, J.; Xie, Y.; Li, X.; Jiang, H.; Wang, J.; Peng, Z.; Wang, J.; Wang, S.; et al. Hypoxia-induced CREB cooperates MMSET to modify chromatin and promote DKK1 expression in multiple myeloma. Oncogene 2021, 40, 1231–1241.
- Egelhofer, T.A.; Minoda, A.; Klugman, S.; Lee, K.; Kolasinska-Zwierz, P.; Alekseyenko, A.A.; Cheung, M.-S.; Day, D.S.; Gadel, S.; Gorchakov, A.A.; et al. An assessment of histone-modification antibody quality. Nat. Struct. Mol. Biol. 2010, 18, 91–93.
- Byrum, S.D.; Taverna, S.D.; Tackett, A.J. Purification of specific chromatin loci for proteomic analysis. Breast Cancer 2014, 1228, 83–92.
- Sidoli, S.; Bhanu, N.V.; Karch, K.R.; Wang, X.; Garcia, B.A. Complete Workflow for Analysis of Histone Post-translational Modifications Using Bottom-up Mass Spectrometry: From Histone Extraction to Data Analysis. J. Vis. Exp. 2016, e54112.
- Gallego, L.D.; Steger, M.G.; Polyansky, A.A.; Schubert, T.; Zagrovic, B.; Zheng, N.; Clausen, T.; Herzog, F.; Köhler, A. Structural mechanism for the recognition and ubiquitination of a single nucleosome residue by Rad6–Bre1. Proc. Natl. Acad. Sci. USA 2016, 113, 10553–10558.
- Singh, V.; Ram, M.; Kumar, R.; Prasad, R.; Roy, B.K.; Singh, K.K. Phosphorylation: Implications in Cancer. Protein J. 2017, 36, 1–6.
- Han, Z.-J.; Feng, Y.-H.; Gu, B.-H.; Li, Y.-M.; Chen, H. The post-translational modification, SUMOylation, and cancer (Review). Int. J. Oncol. 2018, 52, 1081–1094.
- Mansour, M.A. Ubiquitination: Friend and foe in cancer. Int. J. Biochem. Cell Biol. 2018, 101, 80–93.
- Abramson, H.N. The Multiple Myeloma Drug Pipeline—2018: A Review of Small Molecules and Their Therapeutic Targets. Clin. Lymphoma Myeloma Leuk. 2018, 18, 611–627.
- Salesse, S.; Verfaillie, C.M. BCR/ABL: From molecular mechanisms of leukemia induction to treatment of chronic myelogenous leukemia. Oncogene 2002, 21, 8547–8559.
- Raedler, L.A. Jakafi (Ruxolitinib): First FDA-Approved Medication for the Treatment of Patients with Polycythemia Vera. Am. Health Drug Benefits 2015, 8, 75–79.
- Verstovsek, S.; Gotlib, J.; Mesa, R.A.; Vannucchi, A.M.; Kiladjian, J.-J.; Cervantes, F.; Harrison, C.N.; Paquette, R.; Sun, W.; Naim, A.; et al. Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses. J. Hematol. Oncol. 2017, 10, 1–6.
- Levis, M. Midostaurin approved for FLT3-mutated AML. Blood 2017, 129, 3403–3406.
- Stone, R.M.; Manley, P.W.; Larson, R.A.; Capdeville, R. Midostaurin: Its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Adv. 2018, 2, 444–453.
- Dhillon, S. Gilteritinib: First Global Approval. Drugs 2019, 79, 331–339.
- Waller, C.F. Imatinib Mesylate. Methods Mol. Biol. 2018, 212, 1–27.
- Lindauer, M.; Hochhaus, A. Dasatinib. Methods Mol. Biol. 2018, 212, 29–68.
- Sacha, T.; Saglio, G. Nilotinib in the treatment of chronic myeloid leukemia. Futur. Oncol. 2019, 15, 953–965.
- Cortes, J.E.; Gambacorti-Passerini, C.; Deininger, M.W.; Mauro, M.J.; Chuah, C.; Kim, D.-W.; Dyagil, I.; Glushko, N.; Milojkovic, D.; Le Coutre, P.; et al. Bosutinib Versus Imatinib for Newly Diagnosed Chronic Myeloid Leukemia: Results From the Randomized BFORE Trial. J. Clin. Oncol. 2018, 36, 231–237.
- Massaro, F.; Molica, M.; Breccia, M.; Massaro, M.M.A.M.B.F. Ponatinib: A Review of Efficacy and Safety. Curr. Cancer Drug Targets 2018, 18, 847–856.
- Charalambous, A.; Schwarzbich, M.-A.; Witzens-Harig, M. Ibrutinib. Methods Mol. Biol. 2018, 212, 133–168.
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635.
- Robak, P.; Robak, T. Bortezomib for the Treatment of Hematologic Malignancies: 15 Years Later. Drugs R D 2019, 19, 73–92.
- Groen, K.; Van De Donk, N.W.C.J.; Stege, C.; Zweegman, S.; Nijhof, I. Carfilzomib for relapsed and refractory multiple myeloma. Cancer Manag. Res. 2019, ume 11, 2663–2675.
- Richardson, P.G.; Zweegman, S.; O’Donnell, E.K.; Laubach, J.P.; Raje, N.; Voorhees, P.; Ferrari, R.H.; Skacel, T.; Kumar, S.K.; Lonial, S. Ixazomib for the treatment of multiple myeloma. Expert Opin. Pharmacother. 2018, 19, 1949–1968.
- Tomita, A.; Kiyoi, H.; Naoe, T. Mechanisms of action and resistance to all-trans retinoic acid (ATRA) and arsenic trioxide (As2O3) in acute promyelocytic leukemia. Int. J. Hematol. 2013, 97, 717–725.
- Fink, E.C.; Ebert, B.L. The novel mechanism of lenalidomide activity. Blood 2015, 126, 2366–2369.
- Zhu, Y.X.; Kortuem, K.M.; Stewart, A.K. Molecular mechanism of action of immune-modulatory drugs thalidomide, lenalidomide and pomalidomide in multiple myeloma. Leuk. Lymphoma 2013, 54, 683–687.
- Sivaraj, D.; Green, M.M.; Gasparetto, C. Panobinostat for the management of multiple myeloma. Futur. Oncol. 2017, 13, 477–488.
- Bubna, A.K. Vorinostat—An Overview. Indian J. Derm. 2015, 60, 419.
- Rashidi, A.; Cashen, A.F. Belinostat for the treatment of relapsed or refractory peripheral T-cell lymphoma. Futur. Oncol. 2015, 11, 1659–1664.
- Smolewski, P.; Robak, T. The discovery and development of romidepsin for the treatment of T-cell lymphoma. Expert Opin. Drug Discov. 2017, 12, 1–15.