Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Ralf Weiskirchen.
Lipocalin-2 (LCN2), which is a part of the lipocalin transport protein family, is a protein formally known for its role in iron transport and in inflammatory response.
- lipocalin-2
- hepatocellular carcinoma
- fatty liver disease
1. Introduction
Lipocalin 2 (LCN2), which is a protein encoded by the LCN2 gene on the human chromosome locus 9q34.11 and mouse chromosome 2, was first discovered as a protein associated with neutrophil gelatinase. Hence, its other name is neutrophil gelatinase-associated lipocalin (NGAL). It was purified by Kjeldsen and colleagues who established that its sequence did not match any known human protein but showed a high degree of similarity with the murine 24p3 protein, later proven to be its homologue [1]. Apart from the above names, it is also often referred to as 24p3, oncogenic lipocalin, siderocalin, 25 kDa α2-microglobulin-related protein, and uterocalin, indicating its omnipresence and pleiotropic functions in various tissues.
LCN2 is a part of the lipocalin family of proteins with limited mutual sequence homology but a preserved tertiary barrel structure, consisting of eight-stranded anti-parallel β-barrel enclosed by two α-helices forming a hydrophobic pocket. Their primary role is the transport of small hydrophobic molecules including retinoids, steroids, and drugs [2]. The first role assigned to LCN2 was part of the innate immune response to bacterial infection where LCN2 serves as an iron-containing siderophores sequester [3]. It binds bacterial siderophores, thus, preventing the bacterial strategy of iron uptake, leaving them in need of nutrients and, in that way, reducing the duration of infection. Since it may be presumed based on the variety of names, its roles do not end with iron transport. Quite the opposite, its roles range from implication in cell apoptosis [4], lipid metabolism [5], cell migration [6], tumor invasiveness, and even metastasis [7]. It is even included in the regulation of receptors trafficking. For example, it is responsible for the epidermal growth factor receptor (EGFR) intracellular trafficking mediated by TGF-α [8]. It acts by binding to the intracellular domain of EGFR in late endosomal compartments, thus, inhibiting its lysosomal degradation. Constitutive activation of EGFR is widely known as an instigator of tumor cells proliferation, growth, survival, and migration, which is why it is speculated that the inhibition of LCN2 in this context could serve as a therapeutical target.
LCN2 can exist as either a 25-kDa monomer or a 56-kDa homodimer, which is the most abundant form in healthy individuals, or as a 130-kDa LCN2-matrix metalloproteinase 9 (MMP-9) complex. It has been proposed that dimers are the result of prolonged LCN2 storage in neutrophils that allow dimers to be formed before excretion [9]. This explanation comes from the fact that epithelia-derived LCN2 is primarily composed of monomers since it is being excreted directly, without storage [9,10][9][10]. However, to date, no differences in the pathophysiological functions between the monomer and dimer, except for faster clearance of the monomer from circulation, have been reported [11]. Regarding the LCN2/MMP-9 complex, research suggests that LCN2 can act as a supporting factor for MMP-9, making it less prone to hydrolysis and prolonging its activity [12]. The complex is present only in humans since a mouse homologue lacks Cys-87 residue, which forms a di-sulphide bond with an unidentified cysteine residue in pro-MMP-9. For the same reason, murine models also lack the homodimer form [13]. This fact, which is sometimes neglected, is of the most utter importance since the majority of the LCN2 research is done on murine models and could also account for the discrepancies in the results between the human and murine models presented below. There has been a great review written on the problematics by Bauvois and Susin [14]. One other form has been described lately, known as a 30-kDa isoform, which likely originates from different site-glycosylation [15]. Data are scarce regarding the N-glycosylated form of the LCN2 and, for now, it is only reported in literature that the glycosylation makes the protein more soluble, while it is not required neither for secretion nor exosome targeting [15].
2. Regulation of LCN2
The promoter for LCN2 contains binding sites for nuclear factor-κB (NF-κB) and CCAAT/enhancer-binding protein (C/EBP), which goes in agreement with its expression mostly being regulated by inflammatory pathways [16,17][16][17]. It has been reported that the mitogen-activated protein kinase (MAPK) pathway may cooperate with NF-κB to upregulate the expression of LCN2 [14]. The most profound effect on the expression LCN2 have proven to have the pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α), Interleukin 1β (IL-1β), and Interleukin 6 (IL-6) [2]. The LCN2 gene also contains cis-binding elements for transcription factors such as specificity protein 1 (SP1), polyoma enhancer activator (PEA3), lymphocyte function-associated antigen 1 (LFA-A1), and glucocorticoid receptors (GRs) [11]. Considering that these elements are tissue-specific, their presence allows for LCN2’s varying roles in physiological processes. However, new binding sites and also trans-acting elements have been found [18].
To this day, three LCN2 receptors have been discovered and characterized. One is the endocytic low-density lipoprotein receptor megalin, which binds the iron transporting LCN2 with high affinity and mediates its cellular uptake [19]. Even though megalin is a multi-ligand receptor, its higher-binding affinity toward LCN2 compared to other adipokines has been proven, but not completely explained. For now, it is considered that it results from the net positive charge displayed by LCN2, making it easy to differentiate among other adipokines. Another receptor is the solute carrier family 22, member 17 (SLC22A17), which is also known as the organic cation transporter (BOCT), 24p3R or neutrophil gelatinase-associated lipocalin receptor (NGALR) [20]. While megalin does not distinguish between apo and holo-forms of LCN2, SLC22A17 seems to be able to discriminate between the two and selectively mediate apoptosis and iron uptake in cancer cells [21,22][21][22]. A third receptor identified is the melanocortin-4 receptor that is particularly involved in the control of energy metabolism and expenditure [23]. Given that LCN2 is present in a broader range of tissues and possesses many more functions than can be explained by its binding to only these three receptors, it would be no wonder if some new potential receptors for LCN2 arise in the following years. For the moment, evidence points out that LCN2′s roles are a consequence of its lipophilic ligand transport [24]. For instance, lipocalins are known to transport fatty acids and retinoids, which, once inside the cell, can cause activation of signaling pathways or gene transcription [25]. However, it cannot be excluded that LCN2 possess the ability to induce a direct signal through binding to its specific, still undiscovered, membrane receptor.
3. The Expression of LCN2 in Tissues
LCN2′s expression starts in the foetal stage but is fairly low in an adult, healthy human. According to Human Protein Atlas, LCN2 protein is physiologically expressed in nine tissues of the adult body including kidney, cervix, bone marrow, nasopharynges, bronchus, stomach, spleen salivary gland, and skin. However, its RNA expression exceeds that number. It is typically excreted by neutrophils, basophils, ductal cells, pancreatic endocrine cells, exocrine glandular cells, and urothelial cells [Human Protein Atlas available from http://www.proteinatlas.org; last accessed 3 January 2021]. Importantly, all these cells are either specialized in an immune defence or serve as a first barrier (mucosal and physical) for pathogens confirming its role as a “primary defendant”.
Nonetheless, during pathological conditions, the number of cells secreting LCN2 grows. Due to the multitude of its roles, LCN2 shows significant upregulation in various benign and malignant diseases, non-even related to immunological defence. It has been shown that the upregulation of LCN2 is a common factor upon liver injury caused by factors such as excess alcohol consumption, and hepatectomy following hepatitis B and C virus infection [26]. Knowledge gained about LCN2 in the last decade detects its involvement in non-alcoholic fatty liver disease (NAFLD). This is a condition where hepatocytes serve as a major source of LCN2 [27]. However, the role of LCN2 in the pathology has not yet been fully elucidated, which is why, for this review, we aimed to collect current data on its expression in hepatic malfunction with emphasis on elucidating its dysregulation in non-alcoholic liver pathology and subsequent culmination in hepatocellular carcinoma (HCC).
34. LCN2 in NAFLD Pathophysiology
LCN2 has proven to be a good target to study as a possible biomarker for NAFLD and subsequent liver pathology (Figure 2). It has been proven by independent research teams that its high levels can indicate liver damage [45,46][28][29]. Since it can be detected in bodily fluids such as blood and urine, it can be routinely used as a part of laboratory tests.
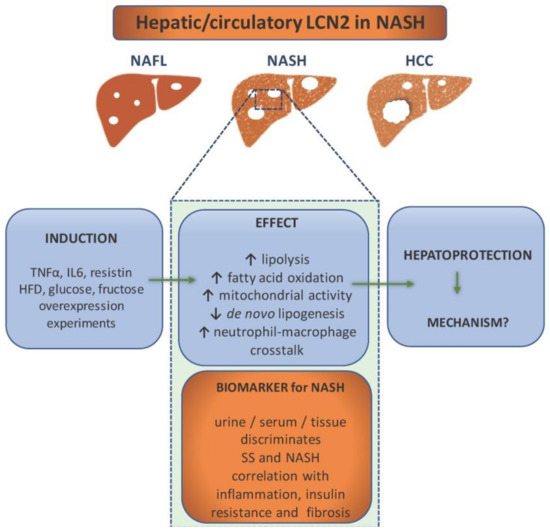
Figure 2. Lipocalin-2 (LCN2) as a component in non-alcoholic steatohepatitis (NASH) pathology and diagnostics. NASH patients show elevated levels of LCN2 in urine, serum, and liver tissue, which makes it a potential biomarker for NASH. It is able to discriminate between simple steatosis (SS) and NASH, while the level of LCN2 correlates with inflammation (e.g., increase of CRP), insulin resistance, and fibrosis. It is induced by proinflammatory cytokines, high fat diet, glucose, and fructose overconsumption. Overexpression experiments have implicated LCN2 in the enhancement of lipolysis, fatty acid oxidation, mitochondrial activity, and immune cells crosstalk. In addition, LCN2 upregulation is associated with reduced de novo lipogenesis. All evidence points out that its role is hepatoprotective. However, the exact mechanisms are ought to be found.
To prove it, one study examined the levels of circulating LCN2 as well as its gene expression in obese women with NAFLD (with either NASH or simple steatosis) and normal liver [47][30]. The research concluded that both gene expression and protein levels were upregulated in obese women with NAFLD. However, gene expression correlated with simple steatosis while protein levels correlated with NASH. The same study showed that treatment with proinflammatory TNF-α, IL6, and resistin causes the upregulation of LCN2 in HepG2 cells [47][30]. Therefore, LCN2 is considered to be a liver’s protective response to inflammation. Another study, done on Chinese subjects, proved that LCN2 serum levels are elevated in patients with NAFLD as opposed to the control and that they highly correlate with both inflammation (C-reactive protein) and insulin resistance [48][31]. The study of Milner and colleagues showed that LCN2 levels correlated with the degree of liver inflammation and the stage of hepatic fibrosis as well as insulin resistance [49][32]. The diagnostic value of serum LCN2 was estimated by Xu and colleagues. Around 500 patients with either NAFLD (steatosis: n = 83, NASH: n = 277) or alcoholic fatty liver disease, together with a healthy control, were recruited for the study. The study showed that three variables, including serum LCN2 level, body mass index (BMI), and low-density lipoprotein (LDL) cholesterol, are positively correlated with NAFLD. The predicted value of LCN2 was calculated by multiple regression analysis, and the predicted value was applied to calculate the cut-off value using receiver operating characteristic (ROC) analysis. The area under the ROC curve of serum LCN2 was 0.987 with a specificity of 100% and a sensitivity of 93.5% for NASH diagnosis, and 0.977 with almost the same specificity and sensitivity for steatosis. Both cases showed a low rate of a false positive and a false negative. These data confirmed high diagnostic value of serum LCN2 in NASH and steatosis. However, although the serum LCN2 levels were compared between NAFL and NASH groups, ROC curve analysis was unable to establish an optimal cut-off value of serum LCN2 levels for distinguishing NASH from NAFL subgroups [50][33]. While all these studies portray LCN2 as a potential biomarker, they have all failed to prove that LCN2 can serve as a tool to differentiate the particular stages of NAFLD. Nonetheless, it would be of high importance to pinpoint its role in the pathogenesis. While, as mentioned above, human studies focus on LCN2 as a biomarker, there had been excessive studies to address LCN2’s role in NAFLD pathogenesis performed both in in vitro and in vivo models.
For instance, classical approach of LCN2 overexpression in hepatocytes done by Xu and colleagues resulted in lipolysis and fatty acid oxidation. Through this study, it was demonstrated that LCN2 overexpression prevents de novo lipogenesis, lipid peroxidation, and apoptosis, thus, preventing steatohepatitis [51][34]. This is one of the many studies that imply that LCN2 regulates liver lipid homeostasis.
A study done by Semba and colleagues aimed to detect if LCN2 can be a factor in the differentiation of simple steatosis and NASH [52][35]. For that purpose, DNA microarray analysis of the liver transcriptomes, RT-qPCR, and immunohistochemistry were done on murine models of simple steatosis (dd Shionogi mice) and NASH (fatty liver Shionogi mice). This study showed that LCN2 is overexpressed in mice with NASH, together with chemokines CXCL1 and CXCL9, while their overexpression was missed in mice with steatosis. What seems to be most interesting, all three proteins have specific localization, likely correlating with their role. LCN2 seems to be localized in hepatocytes and correlates to inflammatory cell clusters due to its implication in neutrophil signaling.
Another study from our group showed a hepatoprotective role of LCN2 both in vivo and in vitro. We fed wild type (WT) and Lcn2-deficient (Lcn2−/−) mice with a methionine and choline deficient (MCD) diet as a nutritional model of NASH. We found that LCN2 maintains lipid homeostasis through, among others, the induction of proteins important for lipid droplet formation named Perilipin 5 (PLIN5). It became clear that depletion of LCN2 or PLIN5 prevented normal intracellular lipid droplet formation in murine models as well as cell lines. The homeostasis was restored after transfection or adenoviral vector infection, which only confirmed the importance of LCN2 in liver protection [53][36]. In another previous investigation, we also performed a comparative analysis of Lcn2−/− and WT, high fat diet-fed mice. We managed to detect the proteins BRIT1/MCPH1, HMGB1, FABP5, and PLIN5 as important factors in LCN2-mediated lipid homeostasis. Besides that, it was shown that LCN2 increased mitochondrial activity, intra-mitochondrial chelatable iron pool, and a peroxisome number, suggesting that it is possible for LCN2 to act as a sensor that measures fat content and adjusts homeostasis by modifying peroxisome numbers and/or mitochondrial activity [54][37].
Another role has been assigned to LCN2 by Ye and colleagues. In their study, they induced NASH by either a high fat, high cholesterol (HFHC) diet or an MCD diet in mice. They could show how LCN2 mediates NASH by promoting neutrophil-macrophage crosstalk via the induction of CXCR2 [55][38]. It was noticed that the infiltration of neutrophils and macrophages was substantially attenuated by genetic depletion of Lcn2, but was augmented by chronic infusion of recombinant LCN2, thus, promoting inflammation. Mice lacking CXCR2 are resistant to LCN2-evoked liver inflammation. Even though the mechanism by which LCN2 may upregulate CXCR2 is still not known, the authors believe that it could be through an NF-κB-dependent mechanism.
As mentioned above, major sources of NAFLD pathogenesis seem to be glucose and fructose lipogenesis-derived FFA. However, a series of recent studies have indicated that sugars can induce NAFLD by means independent of de novo lipogenesis. A study conducted in our group showed how excess fructose leads to hepatic steatosis. However, in this context, fructose appears to directly affect liver homeostasis, thereby, manipulating fat metabolism [56][39]. Fructose might disturb liver homeostasis by promoting lipid uptake into the liver, while LCN2 counteracts lipid uptake. The same study showed there are potential differences between the sexes in LCN2-mediated lipid metabolism. This finding is of the utmost importance because it also shows a potential influence of oestrogens on LCN2-mediated lipid homeostasis. This goes in agreement with another study made by Alwash and colleagues who fed rats a fructose high diet that provoked a gradual increase of the LCN2 level over the course of eight weeks [57][40]. LCN2 levels seemed to correlate both with increased indicators of oxidative stress and mitochondrial dysfunction. Their model suggests a hepatoprotective role of LCN2 since its expression in a rat model seems to be provoked by endotoxins and inflammatory cytokines, as mentioned by many more [46,58][29][41]. Although these studies have managed to portray LCN2 as a hepatoprotective element in the liver, they have not managed to provide an exact mechanism, or mechanisms of its action regarding NAFLD pathology. As a matter of fact, they have not even explained whether LCN2 is a mere consequence of the pathology or its driver.
References
- Kjeldsen, L.; Johnsen, A.H.; Sengeløv, H.; Borregaard, N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J. Biol. Chem. 1993, 268, 10425–10432.
- Asimakopoulou, A.; Weiskirchen, R. Lipocalin 2 in the pathogenesis of fatty liver disease and nonalcoholic steatohepatitis. Clin. Lipidol. 2015, 10, 47–67.
- Goetz, D.H.; Holmes, M.A.; Borregaard, N.; Bluhm, M.E.; Raymond, K.N.; Strong, R.K. The Neutrophil Lipocalin NGAL Is a Bacteriostatic Agent that Interferes with Siderophore-Mediated Iron Acquisition. Mol. Cell 2002, 10, 1033–1043.
- Chien, M.H.; Ying, T.H.; Yang, S.F.; Yu, J.K.; Hsu, C.W.; Hsieh, S.C.; Hsieh, Y.H. Lipocalin-2 Induces Apoptosis in Human Hepatocellular Carcinoma Cells Through Activation of Mitochondria Pathways. Cell Biochem. Biophys. 2012, 64, 177–186.
- Asimakopoulou, A.; Borkham-Kamphorst, E.; Henning, M.; Yagmur, E.; Gassler, N.; Liedtke, C.; Weiskirchen, R. Lipocalin-2 (LCN2) regulates PLIN5 expression and intracellular lipid droplet formation in the liver. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2014, 1841, 1513–1524.
- Chung, I.H.; Chen, C.Y.; Lin, Y.H.; Chi, H.C.; Huang, Y.H.; Tai, P.J.; Lin, K.H. Thyroid hormone-mediated regulation of lipocalin 2 through the Met/FAK pathway in liver cancer. Oncotarget 2015, 6, 15050–15064.
- Wang, Y.P.; Yu, G.R.; Lee, M.J.; Lee, S.Y.; Chu, I.S.; Leem, S.H.; Kim, D.G. Lipocalin-2 negatively modulates the epithelial-to-mesenchymal transition in hepatocellular carcinoma through the epidermal growth factor (TGF-beta1)/Lcn2/Twist1 pathway. Hepatology 2013, 58, 1349–1361.
- Yammine, L.; Zablocki, A.; Baron, W.; Terzi, F.; Gallazzini, M. Lipocalin-2 Regulates Epidermal Growth Factor Receptor Intracellular Trafficking. Cell Rep. 2019, 29, 2067–2077.
- Cai, L.; Rubin, J.; Han, W.; Venge, P.; Xu, S. The Origin of Multiple Molecular Forms in Urine of HNL/NGAL. Clin. J. Am. Soc. Nephrol. 2010, 5, 2229–2235.
- Mårtensson, J.; Bellomo, R. The Rise and Fall of NGAL in Acute Kidney Injury. Blood Purif. 2014, 37, 304–310.
- Chakraborty, S.; Kaur, S.; Guha, S.; Batra, S.K. The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim. Biophys. Acta BBA Rev. Cancer 2012, 1826, 129–169.
- Yan, L.; Borregaard, N.; Kjeldsen, L.; Moses, M.A. The High Molecular Weight Urinary Matrix Metalloproteinase (MMP) Activity Is a Complex of Gelatinase B/MMP-9 and Neutrophil Gelatinase-associated Lipocalin (NGAL)): Modulation of MMP-9 activity by NGAL. J. Biol. Chem. 2001, 276, 37258–37265.
- Holmes, M.A.; Paulsene, W.; Jide, X.; Ratledge, C.; Strong, R.K. Siderocalin (Lcn 2) Also Binds Carboxymycobactins, Potentially Defending against Mycobacterial Infections through Iron Sequestration. Structure 2005, 13, 29–41.
- Bauvois, B.; Susin, S.A. Revisiting Neutrophil Gelatinase-Associated Lipocalin (NGAL) in Cancer: Saint or Sinner? Cancers 2018, 10, 336.
- Borkham-Kamphorst, E.; Van De Leur, E.; Meurer, S.K.; Buhl, E.M.; Weiskirchen, R. N-Glycosylation of Lipocalin 2 Is Not Required for Secretion or Exosome Targeting. Front. Pharmacol. 2018, 9, 426.
- Zhao, P.; Elks, C.M.; Stephens, J.M. The Induction of Lipocalin-2 Protein Expression in Vivo and in Vitro. J. Biol. Chem. 2014, 289, 5960–5969.
- Cortes-Canteli, M.; Luna-Medina, R.; Sanz-SanCristobal, M.; Alvarez-Barrientos, A.; Santos, A.; Perez-Castillo, A. CCAAT/enhancer binding protein β deficiency provides cerebral protection following excitotoxic injury. J. Cell Sci. 2008, 121, 1224–1234.
- Krishnan, K.C.; Sabir, S.; Shum, M.; Meng, Y.; Acín-Pérez, R.; Lang, J.M.; Lusis, A.J. Sex-specific metabolic functions of adipose Lipocalin-2. Mol. Metab. 2019, 30, 30–47.
- Hvidberg, V.; Jacobsen, C.; Strong, R.K.; Cowland, J.B.; Moestrup, S.K.; Borregaard, N. The endocytic receptor megalin binds the iron transporting neutrophil-gelatinase-associated lipocalin with high affinity and mediates its cellular uptake. FEBS Lett. 2005, 579, 773–777.
- Fang, W.K.; Xu, L.Y.; Lu, X.F.; Liao, L.D.; Cai, W.J.; Shen, Z.Y.; Li, E.M. A novel alternative spliced variant of neutrophil gelatinase-associated lipocalin receptor in oesophageal carcinoma cells. Biochem. J. 2007, 403, 297–303.
- Martinez, A.I.C.; Weinhäupl, K.; Lee, W.K.; Wolff, N.A.; Storch, B.; Żerko, S.; Coudevylle, N. Biochemical and Structural Characterization of the Interaction between the Siderocalin NGAL/LCN2 (Neutrophil Gelatinase-associated Lipocalin/Lipocalin 2) and the N-terminal Domain of Its Endocytic Receptor SLC22A17. J. Biol. Chem. 2016, 291, 2917–2930.
- Devireddy, L.R.; Gazin, C.; Zhu, X.; Green, M.R. A Cell-Surface Receptor for Lipocalin 24p3 Selectively Mediates Apoptosis and Iron Uptake. Cell 2005, 123, 1293–1305.
- Mosialou, I.; Shikhel, S.; Liu, J.-M.; Maurizi, A.; Luo, N.; He, Z.; Huang, Y.; Zong, H.; Friedman, R.A.; Barasch, J.; et al. MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature 2017, 543, 385–390.
- Flo, T.H.; Smith, K.D.; Sato, S.; Rodriguez, D.J.; Holmes, M.A.; Strong, R.K.; Aderem, A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 2004, 432, 917–921.
- Bratt, T. Lipocalins and cancer. Biochim. Biophys. Acta BBA Protein Struct. Mol. Enzym. 2000, 1482, 318–326.
- Wieser, V.; Tymoszuk, P.; Adolph, T.E.; Grander, C.; Grabherr, F.; Enrich, B.; Tilg, H. Lipocalin 2 drives neutrophilic inflammation in alcoholic liver disease. J. Hepatol. 2016, 64, 872–880.
- Xu, Y.; Zhu, Y.; Jadhav, K.; Li, Y.; Sun, H.; Yin, L.; Zhang, Y. Lipocalin-2 Protects Against Diet-Induced Nonalcoholic Fatty Liver Disease by Targeting Hepatocytes. Hepatol. Commun. 2019, 3, 763–775.
- Hu, Y.; Xue, J.; Yang, Y.; Zhou, X.; Qin, C.; Zheng, M.; Chen, F. Lipocalin 2 Upregulation Protects Hepatocytes from IL1-β-Induced Stress. Cell. Physiol. Biochem. 2015, 36, 753–762.
- Borkham-Kamphorst, E.; Drews, F.; Weiskirchen, R. Induction of lipocalin-2 expression in acute and chronic experimental liver injury moderated by pro-inflammatory cytokines interleukin-1β through nuclear factor-κB activation. Liver Int. 2011, 31, 656–665.
- Auguet, T.; Terra, X.; Quintero, Y.; Martínez, S.; Manresa, N.; Porras, J.A.; Richart, C. Liver Lipocalin 2 Expression in Severely Obese Women with Non Alcoholic Fatty Liver Disease. Exp. Clin. Endocrinol. Diabetes 2013, 121, 119–124.
- Lu, J.; Lin, L.; Ye, C.; Tao, Q.; Cui, M.; Zheng, S.; Xue, Y. Serum NGAL Is Superior to Cystatin C in Predicting the Prognosis of Acute-on-Chronic Liver Failure. Ann. Hepatol. 2019, 18, 155–164.
- Milner, K.-L.; Van Der Poorten, D.; Xu, A.; Bugianesi, E.; Kench, J.G.; Lam, K.S.; George, J. Adipocyte fatty acid binding protein levels relate to inflammation and fibrosis in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1926–1934.
- Xu, G.; Wang, Y.M.; Ying, M.M.; Chen, S.D.; Li, Z.R.; Ma, H.L.; Zheng, M.H.; Wu, J.; Ding, C.M. Serum Lipocalin-2 is a Potential Biomarker for the Clinical Diagnosis of Nonalcoholic Steatohepatitis. Clin. Mol. Hepatol. 2021. Epub Ahead of Print.
- Xu, H.; Sun, X.; Sun, W.J. Expression and clinical correlation of NGAL and VEGF in endometrial carcinoma. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 632–636.
- Semba, T.; Nishimura, M.; Nishimura, S.; Ohara, O.; Ishige, T.; Ohno, S.; Nomura, F. The FLS (Fatty liver Shionogi) mouse reveals local expressions of lipocalin-2, CXCL1 and CXCL9 in the liver with non-alcoholic steatohepatitis. BMC Gastroenterol. 2013, 13, 1–11.
- Asimakopoulou, A.; Vucur, M.; Luedde, T.; Schneiders, S.; Kalampoka, S.; Weiss, T.S.; Weiskirchen, R. Perilipin 5 and Lipocalin 2 Expression in Hepatocellular Carcinoma. Cancers 2019, 11, 385.
- Asimakopoulou, A.; Fülöp, A.; Borkham-Kamphorst, E.; Van de Leur, E.; Gassler, N.; Berger, T.; Weiskirchen, R. Altered mitochondrial and peroxisomal integrity in lipocalin-2-deficient mice with hepatic steatosis. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2017, 1863, 2093–2110.
- Ye, D.; Yang, K.; Zang, S.; Lin, Z.; Chau, H.T.; Wang, Y.; Wang, Y. Lipocalin-2 mediates non-alcoholic steatohepatitis by promoting neutrophil-macrophage crosstalk via the induction of CXCR2. J. Hepatol. 2016, 65, 988–997.
- Lambertz, J.; Berger, T.; Mak, T.W.; Van Helden, J.; Weiskirchen, R. Lipocalin-2 in Fructose-Induced Fatty Liver Disease. Front. Physiol. 2017, 8, 964.
- Alwahsh, S.M.; Xu, M.; Seyhan, H.A.; Ahmad, S.; Mihm, S.; Ramadori, G.; Schultze, F.C. Diet high in fructose leads to an overexpression of lipocalin-2 in rat fatty liver. World J. Gastroenterol. WJG 2014, 20, 1807.
- Zhang, Y.; Foncea, R.; Deis, J.A.; Guo, H.; Bernlohr, D.A.; Chen, X. Lipocalin 2 Expression and Secretion Is Highly Regulated by Metabolic Stress, Cytokines, and Nutrients in Adipocytes. PLoS ONE 2014, 9, e96997.
More