One of the critical functions of PARP-1 is the response to DNA damage, which plays a pivotal role in DNA repair in cancers. PARP-1 also has widespread functions that are essential for the survival and growth of cancer cells. It regulates oxidative stress in mitochondria through the regulation of superoxide and oxidation. PARP-1 is in charge of regulating mitosis, which is a crucial role in tumorigenesis and remodels histones and chromatin enzymes related to transcriptional regulation, causing alterations in epigenetic markers and chromatin structure. Given the significance of these processes, it can be understood that these processes in cancer cells are at the frontline of the pathogenetic changes required for cancer cell survival, and these contributions can result in malignant transformation.
- pancreatic cancer
- PARP-1
- oxidative stress
- mitosis
1. PARP-1 and Poly-ADP Ribosylation
To interpret the various cellular physiological functions of PARP-1, it is extremely important to understand the biochemical phenomenon defined as poly-ADP-ribosylation (PARylation). PARP-1 consists of a multidomain structure sharing the same catalytic domain that exhibits structural homology to other ADP-ribosyl transferases
. The N-terminal region contains a DNA-binding domain with three zinc fingers and an auto-modification domain. The C-terminal region contains the protein-interacting domain and the catalytic subdomain responsible for ADP-ribosylation reaction
. These domains enable genetic interactions by catalyzing the covalent attachment of poly-ADP-ribose (PAR) polymers on PARP-1 and other acceptor proteins, including histones, DNA repair proteins, transcription factors, and chromatin modulators
. PARP-1 and tankyrases synthesize branched PAR polymers following the cleavage of nicotinamide adenine dinucleotide to nicotinamide and ADP-ribose, and this enzymology reaction is the critical process for PARylation (
)
. These resemble the grab hold of PARP-1 to DNA transcription factors, protein–protein, and protein–nucleic acid interactions
. Although the majority of cellular PARP-1 activity is localized to the nucleus, PAR and PARylated proteins can also be transferred to the cytosol. Therefore, the molecular biological aspects of PARP-1 activity can exert various roles through the pathophysiological outcomes with PARylation
. This rationale can also be accounted for the feature of a unique enzymatic event, namely, PARP-1 activation, with PARylation-mediated cellular physiological changes being associated with the development of cancer
. This process is characterized by alterations at the cellular, genetic, and epigenetic levels comprising a multistep process involving various physiological maintenance and overcoming stress conditions
. PARP-1 and PARylation have been implicated in all these processes, suggesting possible connections between PARP-1 function and tumorigenesis
. In other words, a series of biochemical phenomena associated with PARylation following PARP-1 activation can be a potential theory that can collectively explain the multifactorial process of pancreatic cancer progression (
).
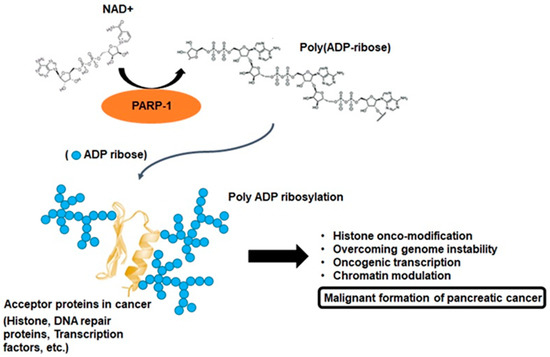
Poly ADP-ribosylation in cancer. PARP-1 branched poly ADP-ribose polymers following the cleavage of nicotinamide adenine dinucleotide (NAD+) to ADP-ribose. PARP-1 enables interactions by catalyzing the covalent attachment of poly ADP-ribose polymers on acceptor proteins, such as histones, DNA repair proteins, transcription factors, and chromatin modulators. This enzymology reaction is known as poly ADP-ribosylation, and this process may be important for pancreatic cancer malignant formation.
2. Multifactorial Functions of PARP-1 in Cancer Progression
2.1. Oxidative Stress and PARP-1
Oxidative phosphorylation is the basic metabolism responsible for the process of generating energy in the mitochondria, and as a result of this metabolism, essential compounds are also synthesized that are beneficial for the cell to survive
. However, the result of this metabolism also causes the cells to undergo oxidative stress due to the production of reactive species following the activation of mitochondrial enzymes
. Reactive oxygen species (ROS) account for a high proportion among these reactive species within the cells, and low concentration levels (<5%) of these species can be involved in benign metabolic events such as cell signaling, enzyme activation, and gene expression
. However, the loss of equilibrium between ROS and endogenous antioxidant species induces intracellular oxidative stress and alteration and damage to several intracellular molecules, including DNA, RNA, lipids, and proteins, even leading to the progression of apoptosis
. Nevertheless, cancer cells can rather function positively under higher levels of oxidative stress conditions, which indicates the possibility that unique adaptive responses to oxidative stress would be an essential factor contributing to malignant transformation, including DNA damage, cell–cell adhesion, and signaling for sustained cancer cell survival
. Regulating the balance or imbalance of intracellular ROS to maintain cellular functions is a well-established hallmark of pancreatic cancer cells. Maintaining a moderately higher level of ROS production than that in normal cells in the tumor microenvironment can act as a signaling molecule to promote the mutation of genomic DNA or enhance the proliferation of pancreatic cancer cells
. On the one hand, excess ROS can cause irreversible oxidative damage, induce cell death through apoptosis, necrosis, and autophagy, and render pancreatic cancer cells susceptible to extracellular turbulences such as chemotherapy and radiation therapy
. Under such a critical environment, the functioning of the antioxidant program protects the pancreatic cancer cells from irreversible oxidative damage. The antioxidant program is driven by defense through enzymatic antioxidants, including the detoxification of secondary metabolites, as well as the direct removal of the electrophile itself
. Interestingly, PARP-1 controls a core role in the areas of such active utilization of oxidative stress conditions similar to a double-edged sword.
Antioxidant enzymes are under the transcriptional control of nuclear factor erythroid-related factor 2 (NRF2), a basic leucine zipper protein, and respond to physiological changes between intracellular redox actions for maintaining cellular homeostasis
. Because PARP-1 promotes the interaction of antioxidant response elements with NRF2 and NRF2-partners, their interaction with PARP-1 enables the entire transcriptional activity of NRF2
. Another counteracting mechanism responsible for oxidant-involving PARP-1 is denoted by its interaction with hypoxia-inducing factor 1-alpha (HIF1-α), which undergoes activation during hypoxia-triggered redox imbalance
. PARP-1 forms a co-transcriptional activator with HIF1-α and enables the expression of the genes required for the maintenance of glutathione homeostasis, such as heme oxygenase-1 and glucose transporter 1, controlled in a PARP-1-dependent manner in a hypoxic response element promoter region
. Furthermore, recent studies have reported that the signaling on ROS is dependent on PARP-1 activation related to the regulation of protein kinase B (AKT)
. Cancer cells with KRAS mutation display constitutive activation of the AKT pathways, resulting in ROS production
. As mentioned above, low concentration levels of ROS can be involved in benign metabolic events, but excessive ROS induces intracellular oxidative stress, such as damage to several intracellular molecules, leading to apoptosis
. The cascade in the AKT pathway, phosphatidylinositol 3, acts as a redox sensor and phosphorylates AKT to induce the active form, and AKT stimulates oxidative metabolism and forkhead box O-dependent catalase inhibition, contributing to the accumulation of hydrogen peroxide, however, PARP-1 activation and PAR synthesis inhibit the mTOR complex 1 signaling pathway and possibly modulate the mTOR complex 2, resulting in AKT downregulation
. The increase in the levels of oxidants enhances the expression of the extracellular signal-regulated kinase (ERK) followed by the regulation of oxidant levels in the cells
. PARP-1 involves the transcription of ERK and mitogen-activated protein kinase phosphatase 1 (MKP-1) in these signaling events, thereby regulating mitogen-activated protein kinase (MAPK)
. Its regulation for the downregulation of MKP-1 blocks the dephosphorylation of the tyrosine and threonine residues of MAPK, which are activated upon acute oxidant exposure
. In other words, PARP-1 is involved in blocking the dephosphorylation of the tyrosine and threonine residues in MAPK, thus contributing to the induction of sustained survival signals even under the increase in ROS (
)
.
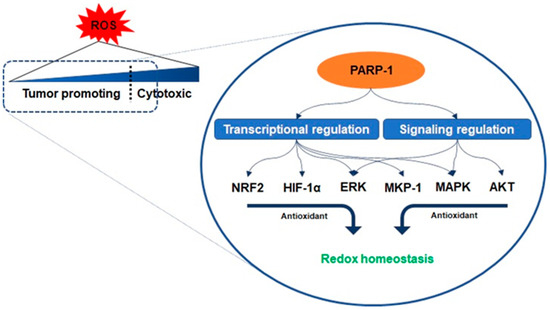
Regulation of redox homeostasis by PARP-1. The activity of PARP-1 plays an important role in the adaptive response of cancer cells to oxidative stress. PARP-1 promotes the interaction of antioxidant response elements with nuclear factor erythroid-related factor 2 (NRF2) and interacts with hypoxia-inducing factor 1-alpha (HIF1-α). PARP-1 activation and poly ADP ribose synthesis possibly modulate the mTOR complex 2, resulting in AKT downregulation to inhibit oxidants’ accumulation. To regulate oxidants level, PARP-1 is in charge of the transcription of the extracellular signal-regulated kinase (ERK) and mitogen-activated protein kinase phosphatase 1 (MKP-1) related to mitogen-activated protein kinase (MAPK) regulation.
2.2. Genomic Instability and PARP-1
Maintenance of genomic stability is important for cellular integrity in normal cells to prevent mutation by endogenous genotoxic stresses or exogenous carcinogenic insults. In contrast, carcinogenesis progresses under genomic alterations in cells accompanied by the selection of aggressive subclones in this process. These conditions provide growth benefits to cancer cells under a genetically unstable state and result in select malignant formation
. Tumor progression requires an increase in genetic alterations and downregulation of DNA damage surveillance mechanisms to achieve uncontrolled proliferation for aggressive growth
. The insights provided through studies on oncogenes could indicate that long-lasting stress from excessive replication promotes genomic instability with sustained damage to DNA
. In pancreatic cancer, silencing or detrimental mutations in the key gene responsible for DNA damage response or tumor suppressor genes, such as breast cancer susceptibility gene (BRCA), ataxia telangiectasia mutated (ATM), and p53, have been associated with increased genomic instability
. BRCA mutations are exposed susceptible to intrinsic or extrinsic stress because of the lack of DNA repair response, and a mutation in the ATM gene causes increased genetic alterations such as deletion or insertion of new nucleotides and chromosomal translocations
. The major tumor suppressor gene p53 can contribute to high genomic instability following the loss of activity
. In other words, the result of genomic instability is a continuing genetic rearrangement throughout the incidence and progression of pancreatic cancer, and various genetic changes have been observed at different loci under metastatic characteristics
. This indicates the possibility that they possess a variety of cancer properties with resistance, even in the same metastatic lesions from the same parental clone.
Genetic instability is generated by excessive replication stress, and either directly or indirectly, it leads to DNA damage initiated by single-stranded DNA (ssDNA) gaps or double-stranded DNA (dsDNA) breaks, followed by the generation of the DNA repair system
. Homologous recombination is a DNA metabolic process found in most types of cells, providing a form of repair targeting complex DNA damage, cross-linking between ssDNA gap and dsDNA break
. DNA damage response gene-mutated pancreatic cancer cells drive a unique DNA repair system that creates specific genotypic and phenotypic features known as BRCAness. The patterns of BRCAness can provide genetic plasticity, and a variety of molecular behaviors and such genotypes are involved in the process of constructive genomic rearrangement, which can directly alter the genomic structure and molecular properties of pancreatic cancer cells
. Another feature that is not directly involved in the DNA repair process of homologous recombination in pancreatic cancer is PARP-1 activation
. As described in the previous section, PARP-1 is a crucial nuclear enzyme of cellular homeostasis that modifies several nuclear proteins by PARylation
. One of the key features of PARP-1 is to repair ssDNA breaks in response to DNA damage by targeting histone core and the linker histone proteins in the nucleus
. Although the detailed roles of histone modifications with PARP-1 need to be elucidated, previous studies have suggested that PARP-1 can facilitate DNA repair by maintaining the open form of chromatin structures
. A specific serine group-bound ADP-ribose depends on a protein known as histone PARylation factor 1, which has been identified as a key protein controlling the DNA damage-induced PARylation and is available to the adaptation to genomic instability
. As PARP-1 constantly recruits elements for DNA repair via PARylation on multiple receptor regions under genomic instability, histone PARylation factor 1 also has an essential role in the regulation of excessive PARP-1 transformation to evade cell death by apoptosis-inducing factors
. In other words, since the abnormal growth of pancreatic cancer constantly induces DNA damage, leading to genomic instability, the PARP-1 activity and PARylation play a major role in the adaptation to genomic instability in pancreatic cancer (
).
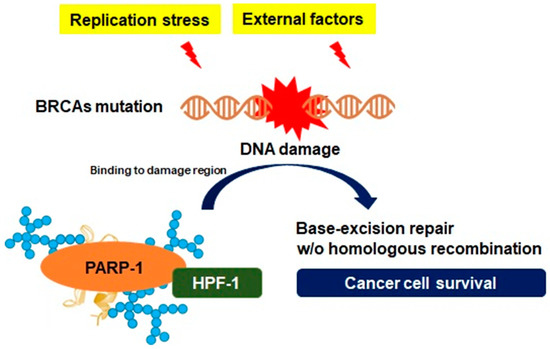
Adaptation of genomic instability depending on PARP-1. Silencing or detrimental mutations in the DNA damage response genes, such as the breast cancer susceptibility gene (BRCA), are associated with genomic instability to achieve uncontrolled proliferation for aggressive growth in cancer cells. However, such an environment could be an unfavorable condition for cancer cell survival, where persistent DNA damage is induced. However, PARP-1 can facilitate DNA repair in response to replication stress or external factors by maintaining the open form of chromatin structures following PAR synthesis on acceptor proteins, such as histone PARylation factor 1 (HPF-1), which is available to the adaptation to genomic instability.
2.3. Mitosis Regulation
Cell division in eukaryotes is a biological process required for the generation of progeny cells, and somatic cell proliferation is controlled via mitosis
. Mitosis comprises four phases within a large frame. Prophase involves mobilization of nuclear fission, chromosomal condensation, centrosome separation, and recruitment of mitotic checkpoint proteins. The cell division then follows metaphase and anaphase and ends with telophase, which completes the processes of chromosomal atrophy and nuclear envelope reassembly around polar chromosomes
. All these processes comprise a sequence to efficiently eliminate errors during mitosis, and accuracy and efficiency are maintained by appropriate regulation of the expression and function of mitotic proteins. This activity is regulated by the mechanisms of phosphorylation and ubiquitination associated with post-translational modification of the gene
. However, the occurrence of a defect can induce abnormal mitosis linking with genetic instability, which can be considered as a hallmark of cancer formation
. The major regulatory function of mitosis depends on the balanced level of the spindle assembly checkpoint protein. Modified expression of the spindle assembly checkpoint protein has been reported in a variety of cancers, and these defects often rely on aneuploidy in normal cells
. Furthermore, defects in chromosome segregation during mitosis can lead to aneuploidy and contribute to genomic instability
. Recent studies have identified mutations for modulators that are important in encoding the protein subunits of the segregation complex in many aneuploidy cancer cells
. Defects in several of these molecular components or upstream regulation of irregulated chromosome numbers and cellular proteins that control poly-polarity, such as the checkpoint kinase and cyclin family proteins, can upregulate the proliferation of cancer cells
.
Recent studies have disclosed an association between the upregulation of PARP-1 activity and mitosis. It has been observed that PARP-1 activity regulates the function of the centrosome to control anaphase and spindle assembly checkpoints
. Moreover, PARylation of mitotic chromatin can contribute toward serving as an indicator of epigenetic regulation at the site of transcription initiation required for transcription reactivation following mitosis
. The centrosome is the primary site of microtubule nucleation required for the formation of new microtubules through the assembly of the mitotic spindle. Therefore, centrosome dysfunction impairs chromosome segregation, promoting aneuploidy and inducing cancerous transformation
. Although the substrate of PARP-1 involved in the regulation of centrosome function has not been characterized, the association between PARP-1 and the centrosome throughout the cell cycle has been inferred by research via PARP-1 inhibition
. It may result from excessive PARylation on the tumor suppressor gene p53, one of the PARP-1 substrates known to regulate centrosome redundancy
. PARP-1 is also accumulated in centrosome chromatin during mitotic metaphase and is dissociated in anaphase following the interaction with centrosome proteins (
)
. Therefore, depletion of PARP-1 causes incomplete synapsis of homologous chromosomes and deficient sister chromatid cohesion
. PARP-1 activity can modify spindle assembly checkpoints through the degradation of cyclin B1 and the downregulation of cyclin-dependent 1 kinase activity, highlighting the point where excessive levels of PARylation have a critical role in the regulation of spindle assembly checkpoints, leading to aneuploidy
. These activities enable driving the tumorigenesis of pancreatic cancer cells via aneuploidy-induced delaminating properties and represent the characteristic of overcoming replication stress
.
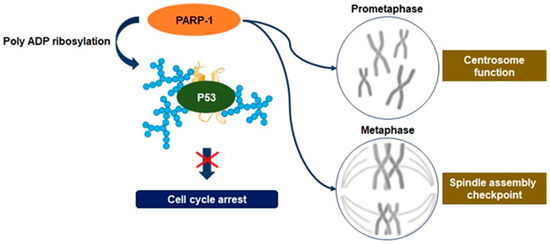
Regulation of mitosis by PARP-1. Defects in chromosome segregation during mitosis can lead to many aneuploidies, and upstream regulation of irregulated chromosome numbers can upregulate the proliferation of cancer cells. PARP-1 regulates the function of the centrosome to control spindle assembly checkpoints in cancer cells, and it may result from excessive poly ADP ribosylation on the tumor suppressor gene p53. PARP-1 is accumulated in centrosome chromatin during mitotic metaphase and can modify spindle assembly checkpoints, leading to aneuploidy. These activities have a role in promoting tumorigenesis of pancreatic cancer cells via aneuploidy-induced delaminating properties.
2.4. Transcriptional Regulation
As genetic dysregulation is one of the primary features of cancer, it is important to gain insights into the specific transcription process involved in the pathogenesis in the cancers. Cutting-edge genomic sequencing conducted to date has allowed us to identify key mutations that affect the transcriptional components and also made it possible to help understand cancer progression by transcriptional regulation. The majority of cancer cells have a feature of constantly maintaining a tumor microenvironment that is favorable for survival through the modification of gene regulators or mutations in signaling factors that are converged on transcriptional regulations
. Genetic variations in cancer can be influenced by changes in proteins that participate in any stage of transcriptional regulation, including transcription factors, co-factors, and promoters
. The final result of these alterations, especially the most profound issues with gene expression, is to contribute to the formation of malignancies
.
Pancreatic cancer can be influenced by a variety of transcriptional regulations, and its activation or inactivation by mutations in transcription factors contributes to this function. The members of the microphthalmia-related transcription factor, transcription factors E3 and EB, regulate the expression of genes associated with high lysosomal activity required for the growth of pancreatic cancer
. Pancreatic duodenum homeobox protein 1 plays a vital role in regulating the early stages of pancreatic development. The pancreas transcription factor 1 subunit-alpha is an essential element that plays an essential role in pancreatic cancer cell differentiation for the early development of pancreatic cancer
. Nuclear receptor subfamily 5 group A member 2, known as liver receptor homolog 1, is a key molecule for the development of pancreatic cancer and is a direct downstream target of pancreatic duodenum homeobox protein 1
. Hepatocyte nuclear factors, also known as transcription factor 1, belong to the homeobox family of proteins that play vital roles in the development of beta cells during pancreatic tissue formation
. All these transcription factors can create a network for interactions that regulate the development of pancreatic cancer. However, before referring to the action of such transcription factors, histone modifications or chromatin remodeling should be prioritized and interactions with enzymes such as the nucleosome can be emphasized essentially. Obviously, PARP-1 not only enables the interaction with nucleosome- and chromatin-related proteins, including transcription factors and components, but it also enables direct communication with DNA
. Therefore, it can be stated that PARP-1 can also be considered as an important marker for identifying the critical relationship between transcription regulation and the development of pancreatic cancer.
PARP-1 is a DNA-dependent ADP-ribosyl transferase that is confined to the nucleus and is frequently associated with chromatin
. The role of PARP-1 activity is not only bound to damaged DNA or other nuclear proteins, but it also includes post-translational modifications by PARylation
. Therefore, it has been demonstrated that PARP-1 accomplishes multiple roles in transcriptional regulation in cancer cells, including properties closely related to genome maintenance, such as DNA repair
. PARP-1 can alter the structure of nucleosomes and chromatin to a form that affects chromatin structure by binding to nucleosomes, compressing chromatin, and inducing PARylation in histones
. Moreover, PARP-1 is localized to the promoter of the transcribed genes and promotes transcriptional activity by interrupting histone binding. Because the transcriptional regulation function of PARP-1 does not generally require co-activity with other enzymes, various functions such as enhancer-binding, regulation of chromatin structure, and transcription factor regulation can independently progress
. The binding of PARP-1 can consistently demonstrate transcriptional regulation, positively or negatively, through several mechanisms, and appear differently according to the demands of the tumor microenvironment (mostly favorable directions for survival). PARP-1 can also immediately regulate the sequence-specific transcription factors that are highly relevant for malignant tumor formation. It constitutes a transcriptional inhibitory complex with p53, and in this context, PARylation can result in the mobilization of histone deacetylases for p53 suppression, leading to increased expression of oncogenes associated with disease progression and metastasis
. PARP-1-dependent interruption of metastasis-associated protein 1 gene expression results in enhanced levels of hypoxia-mediated oncogenes, such as HIF-1α and hexokinase, indicating that the direct transcriptional downregulation of p53 with PARP-1 upregulates cancer-related gene expression and phenotypes
. In other words, the mechanism of transcriptional regulation by PARP-1 in cancer cells can be considered as an important function that affects chromatin remodeling, regulation of tumor suppressor or oncogene expression, metastasis, and cancer cell survival (
). In particular, the p53 gene undergoes deletion in almost 90% of patients with pancreatic cancer, and approximately 60–70% of patients have point mutations that inactivate the remaining gene
. Hence, it is also an important research area to focus on the transcriptional regulation by PARP-1 in pancreatic cancer cells.
References
- Karlberg, T.; Thorsell, A.G.; Kallas, A.; Schuler, H. Crystal structure of human ADP-ribose transferase ARTD15/PARP16 reveals a novel putative regulatory domain. J. Biol. Chem. 2012, 287, 24077–24081.
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-ribosyl)ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019, 47, 3811–3827.
- Marti, J.M.; Fernandez-Cortes, M.; Serrano-Saenz, S.; Zamudio-Martinez, E.; Delgado-Bellido, D.; Garcia-Diaz, A.; Oliver, F.J. The Multifactorial Role of PARP-1 in Tumor Microenvironment. Cancers 2020, 12, 739.
- Pazzaglia, S.; Pioli, C. Multifaceted Role of PARP-1 in DNA Repair and Inflammation: Pathological and Therapeutic Implications in Cancer and Non-Cancer Diseases. Cells 2019, 9, 41.
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958.
- Kamaletdinova, T.; Fanaei-Kahrani, Z.; Wang, Z.Q. The Enigmatic Function of PARP1: From PARylation Activity to PAR Readers. Cells 2019, 8, 1625.
- Solaini, G.; Sgarbi, G.; Baracca, A. Oxidative phosphorylation in cancer cells. Biochim. Biophys Acta 2011, 1807, 534–542.
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462.
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496.
- Zhang, L.; Li, J.; Zong, L.; Chen, X.; Chen, K.; Jiang, Z.; Nan, L.; Li, X.; Li, W.; Shan, T.; et al. Reactive Oxygen Species and Targeted Therapy for Pancreatic Cancer. Oxid. Med. Cell Longev. 2016, 2016, 1616781.
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71.
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426.
- Wu, T.; Wang, X.J.; Tian, W.; Jaramillo, M.C.; Lau, A.; Zhang, D.D. Poly(ADP-ribose) polymerase-1 modulates Nrf2-dependent transcription. Free Radic. Biol. Med. 2014, 67, 69–80.
- Pietrzak, J.; Spickett, C.M.; Ploszaj, T.; Virag, L.; Robaszkiewicz, A. PARP1 promoter links cell cycle progression with adaptation to oxidative environment. Redox. Biol. 2018, 18, 1–5.
- Martinez-Romero, R.; Martinez-Lara, E.; Aguilar-Quesada, R.; Peralta, A.; Oliver, F.J.; Siles, E. PARP-1 modulates deferoxamine-induced HIF-1alpha accumulation through the regulation of nitric oxide and oxidative stress. J. Cell Biochem. 2008, 104, 2248–2260.
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell Longev. 2016, 2016, 4350965.
- Cohen-Armon, M. PARP-1 activation in the ERK signaling pathway. Trends Pharmacol. Sci. 2007, 28, 556–560.
- Durand, N.; Storz, P. Targeting reactive oxygen species in development and progression of pancreatic cancer. Expert Rev. Anticancer Ther. 2017, 17, 19–31.
- Klotz, L.O.; Sanchez-Ramos, C.; Prieto-Arroyo, I.; Urbanek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72.
- Ethier, C.; Tardif, M.; Arul, L.; Poirier, G.G. PARP-1 modulation of mTOR signaling in response to a DNA alkylating agent. PLoS ONE 2012, 7, e47978.
- Racz, B.; Hanto, K.; Tapodi, A.; Solti, I.; Kalman, N.; Jakus, P.; Kovacs, K.; Debreceni, B.; Gallyas, F., Jr.; Sumegi, B. Regulation of MKP-1 expression and MAPK activation by PARP-1 in oxidative stress: A new mechanism for the cytoplasmic effect of PARP-1 activation. Free Radic. Biol. Med. 2010, 49, 1978–1988.
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628.
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42.
- Gorodetska, I.; Kozeretska, I.; Dubrovska, A. BRCA Genes: The Role in Genome Stability, Cancer Stemness and Therapy Resistance. J. Cancer 2019, 10, 2109–2127.
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J. Cell Physiol. 2016, 231, 3–14.
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113.
- Perkhofer, L.; Gout, J.; Roger, E.; Kude de Almeida, F.; Baptista Simoes, C.; Wiesmuller, L.; Seufferlein, T.; Kleger, A. DNA damage repair as a target in pancreatic cancer: State-of-the-art and future perspectives. Gut 2020, 70, 606–617.
- Javle, M.; Curtin, N.J. The role of PARP in DNA repair and its therapeutic exploitation. Br. J. Cancer 2011, 105, 1114–1122.
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621.
- Bonfiglio, J.J.; Fontana, P.; Zhang, Q.; Colby, T.; Gibbs-Seymour, I.; Atanassov, I.; Bartlett, E.; Zaja, R.; Ahel, I.; Matic, I. Serine ADP-Ribosylation Depends on HPF1. Mol. Cell 2017, 65, 932–940.e6.
- Smith, A.P.; Gimenez-Abian, J.F.; Clarke, D.J. DNA-damage-independent checkpoints: Yeast and higher eukaryotes. Cell Cycle 2002, 1, 16–33.
- Bassermann, F.; Eichner, R.; Pagano, M. The ubiquitin proteasome system-implications for cell cycle control and the targeted treatment of cancer. Biochim. Biophys. Acta 2014, 1843, 150–162.
- Ganem, N.J.; Pellman, D. Linking abnormal mitosis to the acquisition of DNA damage. J. Cell Biol. 2012, 199, 871–881.
- Rhind, N.; Russell, P. Signaling pathways that regulate cell division. Cold Spring Harb. Perspect. Biol. 2012, 4, a005942.
- Potapova, T.; Gorbsky, G.J. The Consequences of Chromosome Segregation Errors in Mitosis and Meiosis. Biology 2017, 6, 12.
- Simonetti, G.; Bruno, S.; Padella, A.; Tenti, E.; Martinelli, G. Aneuploidy: Cancer strength or vulnerability? Int. J. Cancer 2019, 144, 8–25.
- Slade, D. Mitotic functions of poly(ADP-ribose) polymerases. Biochem. Pharmacol. 2019, 167, 33–43.
- Ossovskaya, V.; Koo, I.C.; Kaldjian, E.P.; Alvares, C.; Sherman, B.M. Upregulation of Poly (ADP-Ribose) Polymerase-1 (PARP1) in Triple-Negative Breast Cancer and Other Primary Human Tumor Types. Genes Cancer 2010, 1, 812–821.
- Ciccarone, F.; Zampieri, M.; Caiafa, P. PARP1 orchestrates epigenetic events setting up chromatin domains. Semin. Cell Dev. Biol. 2017, 63, 123–134.
- Wu, Q.; Li, B.; Liu, L.; Sun, S.; Sun, S. Centrosome dysfunction: A link between senescence and tumor immunity. Signal. Transduct. Target. Ther. 2020, 5, 107.
- Kanai, M.; Tong, W.M.; Sugihara, E.; Wang, Z.Q.; Fukasawa, K.; Miwa, M. Involvement of poly(ADP-Ribose) polymerase 1 and poly(ADP-Ribosyl)ation in regulation of centrosome function. Mol. Cell Biol. 2003, 23, 2451–2462.
- Kukolj, E.; Kaufmann, T.; Dick, A.E.; Zeillinger, R.; Gerlich, D.W.; Slade, D. PARP inhibition causes premature loss of cohesion in cancer cells. Oncotarget 2017, 8, 103931–103951.
- Palazzo, L.; Della Monica, R.; Visconti, R.; Costanzo, V.; Grieco, D. ATM controls proper mitotic spindle structure. Cell Cycle 2014, 13, 1091–1100.
- Qin, S.; Jiang, J.; Lu, Y.; Nice, E.C.; Huang, C.; Zhang, J.; He, W. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal. Transduct. Target. Ther. 2020, 5, 228.
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643.
- Astanina, E.; Bussolino, F.; Doronzo, G. Multifaceted activities of transcription factor EB in cancer onset and progression. Mol. Oncol. 2021, 15, 327–346.
- Annicotte, J.S.; Fayard, E.; Swift, G.H.; Selander, L.; Edlund, H.; Tanaka, T.; Kodama, T.; Schoonjans, K.; Auwerx, J. Pancreatic-duodenal homeobox 1 regulates expression of liver receptor homolog 1 during pancreas development. Mol. Cell Biol. 2003, 23, 6713–6724.
- Nadolny, C.; Dong, X. Liver receptor homolog-1 (LRH-1): A potential therapeutic target for cancer. Cancer Biol. Ther. 2015, 16, 997–1004.
- Fujitani, Y. Transcriptional regulation of pancreas development and beta-cell function [Review]. Endocr. J. 2017, 64, 477–486.
- Vihervaara, A.; Duarte, F.M.; Lis, J.T. Molecular mechanisms driving transcriptional stress responses. Nat. Rev. Genet. 2018, 19, 385–397.
- Schiewer, M.J.; Knudsen, K.E. Transcriptional roles of PARP1 in cancer. Mol. Cancer Res. 2014, 12, 1069–1080.
- Li, X.; Egervari, G.; Wang, Y.; Berger, S.L.; Lu, Z. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat. Rev. Mol. Cell Biol. 2018, 19, 563–578.
- Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A.; Cicenas, J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers 2017, 9, 42.