One of the critical functions of PARP-1 is the response to DNA damage, which plays a pivotal role in DNA repair in cancers. PARP-1 also has widespread functions that are essential for the survival and growth of cancer cells. It regulates oxidative stress in mitochondria through the regulation of superoxide and oxidation. PARP-1 is in charge of regulating mitosis, which is a crucial role in tumorigenesis and remodels histones and chromatin enzymes related to transcriptional regulation, causing alterations in epigenetic markers and chromatin structure. Given the significance of these processes, it can be understood that these processes in cancer cells are at the frontline of the pathogenetic changes required for cancer cell survival, and these contributions can result in malignant transformation.
- pancreatic cancer
- PARP-1
- oxidative stress
- mitosis
1. PARP-1 and Poly-ADP Ribosylation
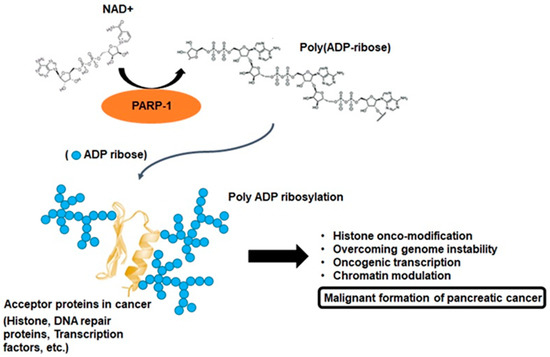
2. Multifactorial Functions of PARP-1 in Cancer Progression
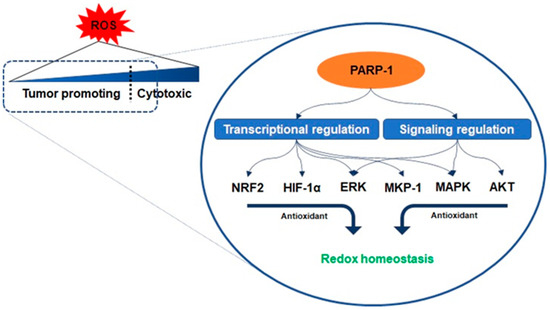
2.1. Oxidative Stress and PARP-1
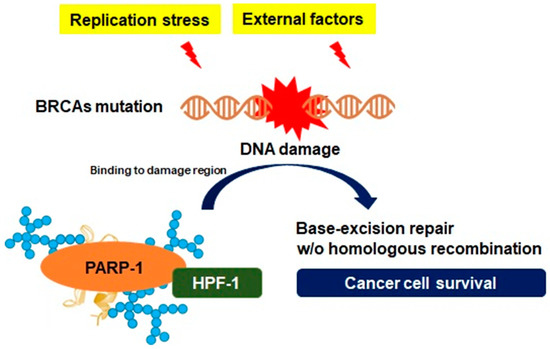
2.2. Genomic Instability and PARP-1
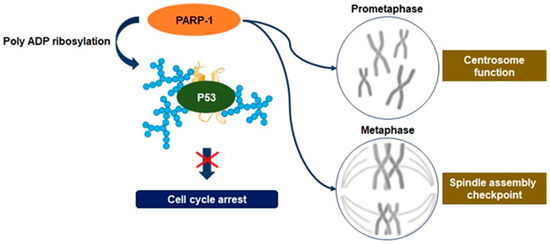
2.3. Mitosis Regulation
2.4. Transcriptional Regulation
References
- Karlberg, T.; Thorsell, A.G.; Kallas, A.; Schuler, H. Crystal structure of human ADP-ribose transferase ARTD15/PARP16 reveals a novel putative regulatory domain. J. Biol. Chem. 2012, 287, 24077–24081.
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-ribosyl)ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019, 47, 3811–3827.
- Marti, J.M.; Fernandez-Cortes, M.; Serrano-Saenz, S.; Zamudio-Martinez, E.; Delgado-Bellido, D.; Garcia-Diaz, A.; Oliver, F.J. The Multifactorial Role of PARP-1 in Tumor Microenvironment. Cancers 2020, 12, 739.
- Pazzaglia, S.; Pioli, C. Multifaceted Role of PARP-1 in DNA Repair and Inflammation: Pathological and Therapeutic Implications in Cancer and Non-Cancer Diseases. Cells 2019, 9, 41.
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958.
- Kamaletdinova, T.; Fanaei-Kahrani, Z.; Wang, Z.Q. The Enigmatic Function of PARP1: From PARylation Activity to PAR Readers. Cells 2019, 8, 1625.
- Solaini, G.; Sgarbi, G.; Baracca, A. Oxidative phosphorylation in cancer cells. Biochim. Biophys Acta 2011, 1807, 534–542.
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462.
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496.
- Zhang, L.; Li, J.; Zong, L.; Chen, X.; Chen, K.; Jiang, Z.; Nan, L.; Li, X.; Li, W.; Shan, T.; et al. Reactive Oxygen Species and Targeted Therapy for Pancreatic Cancer. Oxid. Med. Cell Longev. 2016, 2016, 1616781.
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71.
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426.
- Wu, T.; Wang, X.J.; Tian, W.; Jaramillo, M.C.; Lau, A.; Zhang, D.D. Poly(ADP-ribose) polymerase-1 modulates Nrf2-dependent transcription. Free Radic. Biol. Med. 2014, 67, 69–80.
- Pietrzak, J.; Spickett, C.M.; Ploszaj, T.; Virag, L.; Robaszkiewicz, A. PARP1 promoter links cell cycle progression with adaptation to oxidative environment. Redox. Biol. 2018, 18, 1–5.
- Martinez-Romero, R.; Martinez-Lara, E.; Aguilar-Quesada, R.; Peralta, A.; Oliver, F.J.; Siles, E. PARP-1 modulates deferoxamine-induced HIF-1alpha accumulation through the regulation of nitric oxide and oxidative stress. J. Cell Biochem. 2008, 104, 2248–2260.
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell Longev. 2016, 2016, 4350965.
- Cohen-Armon, M. PARP-1 activation in the ERK signaling pathway. Trends Pharmacol. Sci. 2007, 28, 556–560.
- Durand, N.; Storz, P. Targeting reactive oxygen species in development and progression of pancreatic cancer. Expert Rev. Anticancer Ther. 2017, 17, 19–31.
- Klotz, L.O.; Sanchez-Ramos, C.; Prieto-Arroyo, I.; Urbanek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72.
- Ethier, C.; Tardif, M.; Arul, L.; Poirier, G.G. PARP-1 modulation of mTOR signaling in response to a DNA alkylating agent. PLoS ONE 2012, 7, e47978.
- Racz, B.; Hanto, K.; Tapodi, A.; Solti, I.; Kalman, N.; Jakus, P.; Kovacs, K.; Debreceni, B.; Gallyas, F., Jr.; Sumegi, B. Regulation of MKP-1 expression and MAPK activation by PARP-1 in oxidative stress: A new mechanism for the cytoplasmic effect of PARP-1 activation. Free Radic. Biol. Med. 2010, 49, 1978–1988.
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628.
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42.
- Gorodetska, I.; Kozeretska, I.; Dubrovska, A. BRCA Genes: The Role in Genome Stability, Cancer Stemness and Therapy Resistance. J. Cancer 2019, 10, 2109–2127.
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J. Cell Physiol. 2016, 231, 3–14.
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113.
- Perkhofer, L.; Gout, J.; Roger, E.; Kude de Almeida, F.; Baptista Simoes, C.; Wiesmuller, L.; Seufferlein, T.; Kleger, A. DNA damage repair as a target in pancreatic cancer: State-of-the-art and future perspectives. Gut 2020, 70, 606–617.
- Javle, M.; Curtin, N.J. The role of PARP in DNA repair and its therapeutic exploitation. Br. J. Cancer 2011, 105, 1114–1122.
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621.
- Bonfiglio, J.J.; Fontana, P.; Zhang, Q.; Colby, T.; Gibbs-Seymour, I.; Atanassov, I.; Bartlett, E.; Zaja, R.; Ahel, I.; Matic, I. Serine ADP-Ribosylation Depends on HPF1. Mol. Cell 2017, 65, 932–940.e6.
- Smith, A.P.; Gimenez-Abian, J.F.; Clarke, D.J. DNA-damage-independent checkpoints: Yeast and higher eukaryotes. Cell Cycle 2002, 1, 16–33.
- Bassermann, F.; Eichner, R.; Pagano, M. The ubiquitin proteasome system-implications for cell cycle control and the targeted treatment of cancer. Biochim. Biophys. Acta 2014, 1843, 150–162.
- Ganem, N.J.; Pellman, D. Linking abnormal mitosis to the acquisition of DNA damage. J. Cell Biol. 2012, 199, 871–881.
- Rhind, N.; Russell, P. Signaling pathways that regulate cell division. Cold Spring Harb. Perspect. Biol. 2012, 4, a005942.
- Potapova, T.; Gorbsky, G.J. The Consequences of Chromosome Segregation Errors in Mitosis and Meiosis. Biology 2017, 6, 12.
- Simonetti, G.; Bruno, S.; Padella, A.; Tenti, E.; Martinelli, G. Aneuploidy: Cancer strength or vulnerability? Int. J. Cancer 2019, 144, 8–25.
- Slade, D. Mitotic functions of poly(ADP-ribose) polymerases. Biochem. Pharmacol. 2019, 167, 33–43.
- Ossovskaya, V.; Koo, I.C.; Kaldjian, E.P.; Alvares, C.; Sherman, B.M. Upregulation of Poly (ADP-Ribose) Polymerase-1 (PARP1) in Triple-Negative Breast Cancer and Other Primary Human Tumor Types. Genes Cancer 2010, 1, 812–821.
- Ciccarone, F.; Zampieri, M.; Caiafa, P. PARP1 orchestrates epigenetic events setting up chromatin domains. Semin. Cell Dev. Biol. 2017, 63, 123–134.
- Wu, Q.; Li, B.; Liu, L.; Sun, S.; Sun, S. Centrosome dysfunction: A link between senescence and tumor immunity. Signal. Transduct. Target. Ther. 2020, 5, 107.
- Kanai, M.; Tong, W.M.; Sugihara, E.; Wang, Z.Q.; Fukasawa, K.; Miwa, M. Involvement of poly(ADP-Ribose) polymerase 1 and poly(ADP-Ribosyl)ation in regulation of centrosome function. Mol. Cell Biol. 2003, 23, 2451–2462.
- Kukolj, E.; Kaufmann, T.; Dick, A.E.; Zeillinger, R.; Gerlich, D.W.; Slade, D. PARP inhibition causes premature loss of cohesion in cancer cells. Oncotarget 2017, 8, 103931–103951.
- Palazzo, L.; Della Monica, R.; Visconti, R.; Costanzo, V.; Grieco, D. ATM controls proper mitotic spindle structure. Cell Cycle 2014, 13, 1091–1100.
- Qin, S.; Jiang, J.; Lu, Y.; Nice, E.C.; Huang, C.; Zhang, J.; He, W. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal. Transduct. Target. Ther. 2020, 5, 228.
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643.
- Astanina, E.; Bussolino, F.; Doronzo, G. Multifaceted activities of transcription factor EB in cancer onset and progression. Mol. Oncol. 2021, 15, 327–346.
- Annicotte, J.S.; Fayard, E.; Swift, G.H.; Selander, L.; Edlund, H.; Tanaka, T.; Kodama, T.; Schoonjans, K.; Auwerx, J. Pancreatic-duodenal homeobox 1 regulates expression of liver receptor homolog 1 during pancreas development. Mol. Cell Biol. 2003, 23, 6713–6724.
- Nadolny, C.; Dong, X. Liver receptor homolog-1 (LRH-1): A potential therapeutic target for cancer. Cancer Biol. Ther. 2015, 16, 997–1004.
- Fujitani, Y. Transcriptional regulation of pancreas development and beta-cell function [Review]. Endocr. J. 2017, 64, 477–486.
- Vihervaara, A.; Duarte, F.M.; Lis, J.T. Molecular mechanisms driving transcriptional stress responses. Nat. Rev. Genet. 2018, 19, 385–397.
- Schiewer, M.J.; Knudsen, K.E. Transcriptional roles of PARP1 in cancer. Mol. Cancer Res. 2014, 12, 1069–1080.
- Li, X.; Egervari, G.; Wang, Y.; Berger, S.L.; Lu, Z. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat. Rev. Mol. Cell Biol. 2018, 19, 563–578.
- Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A.; Cicenas, J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers 2017, 9, 42.