Microbiota–gut–brain axis (MGBA) is a bidirectional signaling pathway mediating the interaction of the microbiota, the intestine, and the central nervous system. While the MGBA plays a pivotal role in normal development and physiology of the nervous and gastrointestinal system of the host, its dysfunction has been strongly implicated in neurological disorders, where intestinal dysbiosis and derived metabolites cause barrier permeability defects and elicit local inflammation of the gastrointestinal tract, concomitant with increased pro-inflammatory cytokines, mobilization and infiltration of immune cells into the brain, and the dysregulated activation of the vagus nerve, culminating in neuroinflammation and neuronal dysfunction of the brain and behavioral abnormalities.
- microbiota
- gut–brain axis
- zebrafish
- genetic approach
- in vivo imaging
- gnotobiotic
1. Introduction
The microbiota–gut–brain axis (MGBA) is a bidirectional signaling cascade in which efferent signaling pathways originating from the central nervous system (CNS) regulate the activities of the intestine and the microbiota, while afferent signaling originating from the microbiota and the intestines affects the development and the function of the CNS [1]. The MGBA mainly consists of gut microbiota residing in the intestinal lumen, intestinal cells including enterocytes, enteroendocrine cells (EECs), goblet cells, and neurons and glia in the CNS. Gut microbiota have been shown to be required for normal CNS homeostasis. For example, germ-free (GF) mice have been reported to display hypermyelination in the prefrontal cortex [2] and to have defective microglial maturation and functions [3]. The actions of the MGBA are known to be mediated by metabolites and cytokines that are generated by members of the gut microbiota or released from immune cells and intestinal cells activated by them, or by the streamlined direct connections between the brainstem and intestines via the vagus nerve [4]. Imbalances of the gut microbiota, referred to as dysbiosis, and any associated malfunctions of the MGBA have been implicated in a variety of neurodevelopmental, neuropsychological, and neurodegenerative diseases. These dysbiotic malfunctions have been closely associated with aberrant systemic inflammatory responses and have been shown to culminate in the brain defects that lead to behavioral defects and neuronal dysfunctions [5]. Thus, a more detailed understanding of the underlying mechanisms and physiological roles of the MGBA in the etiopathology in diseases will help to design novel therapeutics based on modulating MGBA activities. For these purposes, the zebrafish has emerged as an excellent animal model system to address the host–microbe interactions for both normal physiology and pathogenesis in vivo.
2. MGBA Pathways
In a pioneering study, GF mice were shown to display exaggerated stress responses and enhanced stress hormone levels that were reversed by the colonization of beneficial bacteria or commensal microbiota, indicating that the gut bacteria play a critical role in the regulation of brain function [6]. Subsequent studies showed that GF mice exhibited hypermotor activity and reduced anxiety, concomitant with changes in gene expression profiles that are important for brain development, and with changes in essential neurotransmitters, specifically in the striatum [7]. The permeability of both the gastrointestinal barrier and the blood–brain barrier (BBB), separating luminal contents from the intestines or blood vessel contents from the brain, respectively, should be tightly regulated by the host to maintain their functional integrities, as the unchecked translocation of bacterial components and metabolites could elicit detrimental inflammation in both the intestine and the brain. Pro-inflammatory cytokines, such as tumor necrosis factor (TNF) and interferon γ (IFNγ), have been shown to regulate the functions of tight junctions [8,9,10][8][9][10]. The MGBA pathways and important signaling mediators are summarized in Figure 1.
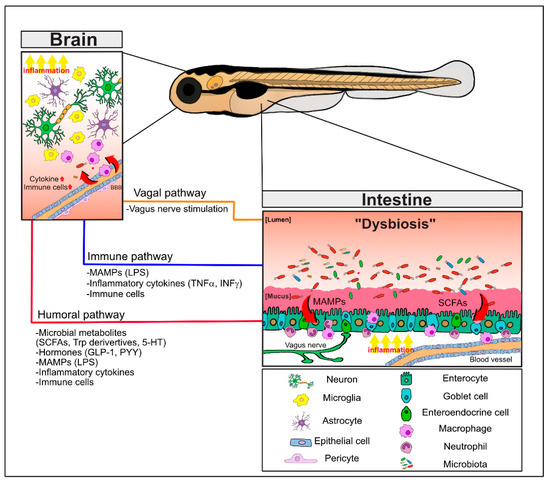
Figure 1. The bidirectional pathways of the microbiota–gut–brain axis (MGBA) involving the microbiota, the intestine, and the brain. Intestinal dysbiosis and derived metabolites in neuropathological conditions induce the barrier permeability defects and local inflammation with increased pro-inflammatory cytokines, MAMPs (e.g., LPS), and activation of immune cells in the gastrointestinal tract. These signaling mediators as well as microbial metabolites (e.g., SCFAs and Trp derivatives) and hormones (e.g., GLP-1, PYY) can distantly affect the brain function via the humoral pathway. In addition, dysregulated regulation of the vagus nerve can also directly modulate the brain function. Together, these MGBA pathways culminate in regulating neuroinflammation and neuronal defects of the brain and behavioral abnormalities. Refer to the text for details. 5-HT; 5-hydroxyltryptamine; LPS, lipopolysaccharides; MAMPs, microbe-associated molecular patterns; SCFAs, short chain fatty acids; Trp; tryptophan.
2.1. Regulation of Brain and Intestinal Permeability by the Microbiota
In GF mice, their intestinal barriers exhibited decreased permeability, with increased expression of several tight junction proteins and immature structural features, which were all reversed after associations with human commensal microbiota [11]. In addition, the composition of gut bacteria can directly affect gut permeability by determining its mucus layer properties [12], and pathogens such as Bacteroides fragilis and Vibrio cholerae or probiotic strains such as Lactobacillus plantarum can change intestinal permeability by regulating tight junction proteins [13], all indicating critical roles for gut microbiota in the regulation of gut permeability. As a result, the changes in intestinal permeability affected by infections or by exposure to dysbiotic products such as bacterial toxins and metabolites can result in low-level chronic and systemic inflammation, eventually developing into a variety of diseases [10,14][10][14].
The BBB in several brain areas is also regulated by the gut microbiota. However, in contrast to the intestinal barrier, it became more permeable in GF mice, with reduced expressions of endothelial tight junction proteins, and these were rescued after associating with the commensal microbiota [15]. Exposure to short chain fatty acid (SCFA)-producing bacteria or to butyrate treatment also reversed BBB permeability defects via the upregulation of tight junction proteins, indicating the crucial involvement of bacterial SCFAs in BBB permeability [15] as well as gut permeability [16].
2.2. Neuronal Communication via the Vagus Nerve
Both the expression of gamma-aminobutyric acid (GABA) receptors in the brain and the improvements in anxiety and depression behaviors regulated by probiotic lactic acid bacteria in mice have been shown to be mediated by the vagus nerve; this has been confirmed by vagotomy where the vagus nerve that connects the brain and the intestine is surgically disconnected [17]. In addition, a subset of intestinal EECs, termed neuropods, has been identified as forming physical synapses with the vagus nerve as part of a neuroepithelial circuit that can sense intestinal signals and convey that information directly to the brain [18]. A neuronal circuit via the vagal nerve has also been described in zebrafish (see below).
2.3. Immune Cell Infiltration into the Brain and Inflammatory Cytokines
Several types of innate immune cells are found in the brain, including residential microglia, perivascular macrophages, and infiltrating macrophages. The infiltrating macrophages of the brain are derived from peripheral bone marrow pro-inflammatory monocytes that transmigrate across the BBB and differentiate into activated macrophages [19]. Neuroinflammation can provoke this type of infiltration, the hallmarks of which are activated microglia as well as the increased expressions of pro-inflammatory cytokines and chemokines such as interleukin-1β (IL-1β), IL-6, IL-17, TNFα, and monocyte chemoattractant protein 1 (MCP1, also called CCL2) in the inflamed brain [20,21][20][21]. The selective depletion of infiltrating monocytes worsened the Aβ load in the Alzheimer’s disease (AD) brain [22] and infiltrating macrophages were shown to have a higher phagocytic capacity against toxic molecules (i.e., Aβ) in AD compared to residential microglia [20[20][21],21], indicating that monocyte infiltration plays a critical role in Aβ clearance. Neutrophils have also been shown to infiltrate into the brain using mouse AD models, and were attracted to amyloid plaques in an LFA-1 integrin-dependent manner [23,24][23][24]. Neutrophil depletion, or LFA-1 blockade, was reported to reduce the neuropathological phenotypes and behavioral deficits in AD mouse models [24]. Furthermore, adaptive immune cells (e.g., clonally expanded CD8+ T cells) have been found in the cerebrospinal fluid (CSF), perivascular regions, and the parenchyma of AD brains [25[25][26],26], although any functional significance and detailed infiltration routes remain to be elucidated.
The neuroinflammation that promotes the infiltration of peripheral immune cells into the brain is likely associated with MGBA function because systemic inflammation elicited by acute infections or by gut dysbiosis, such as inflammatory bowel disease (IBD), has been shown to be strongly associated with neuroinflammatory responses [27]. During dysbiosis, changes in beneficial or pathogenic bacteria and their metabolites (e.g., reduction of Faecalibacterium prausnitzii and its metabolite butyrate) may affect the integrity of intestinal barriers by regulating tight junction protein expressions [28,29][28][29], eventually resulting in the mobilization of activated immune cells in the intestine and the increased expression of inflammatory cytokines [14]. Infiltration of peripheral immune cells into the brain may be further facilitated by systemic inflammation because BBB permeability is also compromised as a result [10]. Consistent with this process, gut dysbiosis was closely associated with the infiltration of pro-inflammatory T helper 1 (Th1) cells into mouse brain using the 5XFAD model and with microglia differentiating into the pro-inflammatory M1 type [30].
The infiltration of immune cells into the brain due to intestinal inflammation has also been observed using a non-mammalian AD model. In a Drosophila model overexpressing amyloid β42 (Aβ42), a non-lethal enterobacterial infection promoted the infiltration of hemocytes (invertebrate phagocytic immune cells) and induced neurodegeneration via the TNF/JNK pathway due to enhanced oxidative stress [31].
2.4. MGBA Metabolites: SCFAs and Tryptophan Derivatives, Including Serotonin
Microbial metabolites have been proposed to be the mediators that link microbiota changes to host metabolism in both normal physiology and disease [32]. The effects of these metabolites on the CNS are via the activation of nerves innervating the intestine, activation/mobilization of immune cells residing in the intestine, or by activating the release of molecules (i.e., endocrine peptides and cytokines) via humoral pathways [33]. The SCFAs and tryptophan/serotonin (also known as 5-hydroxytryptamine or 5-HT) are among the best characterized microbial metabolites that are important for MGBA function and are discussed below.
3. MGBA-Associated Neurological Disorders
Not only does it regulate the normal physiology in the brain, but the gut microbiota is also implicated in pathological conditions of the nervous system via the various gut–brain interactions described above. Acute systemic inflammation due to pathogen infections of the intestine and low-grade chronic inflammation due to intestinal dysbiosis and its derived products of bacterial toxins, metabolites, and cytokines [14] are known to be closely associated with the progression of neurological disorders [59][34]. Below, the diverse roles of the gut microbiota in autism spectrum disorder (ASD), representing a neurodevelopmental disorder, and AD, representing a neurodegenerative disease, will be discussed in detail.
3.1. Autism. Spectrum Disorder: A Neurodevelopmental Disorder
ASD represents a group of neurodevelopmental disorders often diagnosed in childhood and characterized by impaired social interactions and repetitive/restrictive behaviors that are frequently accompanied by intellectual disabilities and epilepsy [60][35]. The etiology of ASD is both complex and heterogeneous, as ASD encompasses a range of symptoms with genetic and environmental factors contributing to its pathogenesis. Large-scale exomic and genomic sequencing of ASD patients has identified a number of candidate genes implicated in neuronal connections, synaptic function, chromatin remodeling/transcription, and RNA splicing [61,62,63][36][37][38]. Notably, a gestational inflammatory environment, such as an infection during pregnancy, can dramatically affect neurodevelopmental and autistic outcomes [64][39]. Based on this idea, inflammatory challenges during pregnancy in animal models, such as viral mimetic poly polyinosinic:polycytidylic acid (I:C) exposure, have been used successfully to establish ASD models for mechanism studies, such as the maternal immune activation mouse model (e.g., [65][40]).
For the pathobiology of ASD, neurodevelopmental defects in the brain have been the main focus due to both the implication of ASD-susceptible genes in neuronal function and the known morphological abnormalities of ASD brains [66,67][41][42]. However, it is also well known that ASD patients have elevated systemic and brain inflammation indicated by increased inflammatory cytokines (e.g., IL6, MCP1, IFN1, and TNFα), activated microglia, and autoantibodies [64,68,69][39][43][44].
Considering that such abnormal inflammatory regulation is thought to be closely linked to the gut microbiota, dysbiosis has been suggested as a main factor responsible for ASD etiology, and this is also consistent with the comorbid gastrointestinal disturbances of ASD patients and their vulnerability to infections [60,70,71,72][35][45][46][47]. In support of this idea, GF animals have been reported to exhibit ASD-like traits (e.g., social avoidance and repetitive behaviors) which were rescued by association of the conventional microbiota [73][48], and both human patients and several ASD mouse models have been shown to have gut dysbiosis [65,74,75,76][40][49][50][51]. In ASD dysbiosis, altered microbial populations and decreased diversity have both been documented, with Clostridium spp. proposed to be the main culprit by producing neurotoxins that are transported via the vagus nerve (e.g., [77][52]), but consistent changes in microbiota profiles have yet to be identified [75][50]. The most direct evidence for a causal relationship between the gut microbiota and ASD has come from recent “humanized” ASD mouse models: the offspring from GF mice harboring a gut microbiota from ASD-patients exhibited ASD-like phenotypes, and further transcriptomic analyses identified aberrant alternative splicing of ASD-risk genes similar to that seen in human ASD brains [78][53]. In support of a microbiota role (and their metabolites) for such ASD-related phenotypes, ASD model mice were rescued by a variety of approaches, including the use of prebiotics/probiotics (Lactobacillus reuteri), antibiotics (vancomycin), metabolites (GABA agonists taurine and 5-aminovaleric acid), specific diets (a ketogenic diet), and microbial transfer therapy [74,78,79,80][49][53][54][55].
The etiology of ASD, especially as it relates to GI problems, is postulated to begin with increased gut permeability (“leaky gut”) in the offspring due to gut dysbiosis under conditions of elevated inflammation such as during maternal immune activation (MIA) or with a high-fat diet (HFD) [8,65,81][8][40][56]. An increased level of toxic metabolites, such as 4-ethylphenylsulfate (4-EHP) and 5-HT [65[40][57],82], or a decreased level of beneficial metabolites, such as butyrate during dysbiosis [28], may also impact gut permeability by modulating the expression of tight junction proteins in the intestine [75][50]. Such a dysfunctional intestinal permeability may allow for the unrestricted entry of dietary components, bacterial components, and metabolites that can elicit increased local intestinal inflammation that is coupled with increased plasma levels of pro-inflammatory cytokines (e.g., IL-6), resulting in systemic inflammation [14] or the permeability changes may directly disturb vagus nerve-dependent signaling to the brain [17]. Notably, IL-6 has also been shown to impair tight junctions by inducing pore-forming claudin-2 [83][58], further worsening gut permeability. It is well established that systemic inflammation can disrupt BBB integrity in neurological disorders [10]. As a result, the compromised BBB may be permissive to peripheral pro-inflammatory cytokines (e.g., IL-6), metabolites (e.g., p-cresol and propionate), or the infiltration of peripheral immune cells in ASD patients [84,85][59][60], with ensuing increased neuroinflammation. Dysregulated neuroinflammation in ASD is known to play a crucial role in its pathogenesis and is mediated by activated microglia and astrocytes that impact dendritic branching, spine density, and neuronal connectivity as well as elevated cytokine expression, finally leading to both cognitive and behavioral abnormalities [86,87][61][62].
Therefore, treating ASD by gut microbiota manipulation may offer more efficient therapeutic approaches which include antibiotics, prebiotics, probiotics, beneficial metabolites, a gluten-free diet, or fecal microbiota transplantation. For example, butyric acid administration is known to ameliorate ASD phenotypes in both mouse models and human patients, presumably by modulating mitochondrial function and neurotransmitter gene expression; interestingly, other SCFAs (e.g., acetate and propionate) did not show any rescue effect; rather, propionate has been used to reversibly induce ASD phenotypes and to generate ASD animal models [75][50]. Furthermore, in several ASD models where mice exhibited gut dysbiosis (e.g., maternal HFD-fed mice, BTBR mice, and shank3 knockout mice) supplementation with L. reuteri was able to rescue the ASD-like social interactions, although this was achieved by vagus nerve stimulation that activated oxytocin release from specific neurons in the ventral tegmental area of the brain, and not by microbial changes [74,88][49][63]. Both gluten-free and ketone-rich diets have been reported to rescue GI abnormalities and ASD symptoms, probably by regulating mitochondria-associated energy metabolism and oxidative stress [89][64] as well as by modulating the gut microbiota [90,91][65][66]. In addition, direct “microbial transfer therapy”, using two weeks of vancomycin treatment followed by eight weeks of fecal microbiota transplantation, has been shown to improve these gut abnormalities and autistic phenotypes and to change the abundance of beneficial bacteria in ASD patients [92][67]. However, as these interventions may often have unwanted side effects such as constipation, and sometimes have conflicting therapeutic effects, more detailed mechanistic studies and optimization protocols are still required for personalized applications.
3.2. Alzheimer’s Disease: A Neurodegeneration Problem
There are two forms of AD: early onset (EOAD, or familial) and late onset (LOAD), with the latter accounting for more than 95% of cases that usually occur after the age of 65. AD is pathologically characterized by the accumulation of amyloid β (Aβ) species and phosphorylated tau protein, leading to the formation of neurotoxic amyloid plaques and neurofibrillary tangles in the brain, respectively, in addition to neuroinflammation, loss of synapses, cognitive impairment, and the eventual loss of neurons [93,94][68][69]. The non-genetic risk factors that are well known for AD include aging, some metabolic disorders, and diets [95[70][71][72],96,97], and both large-scale genome-wide associated studies (GWAS) and whole-genome sequencing of AD patients have identified more than 30 high-risk genetic loci for the disease [98][73].
Previous studies have suggested that these risk factors—aging along with some metabolic syndromes and diets—may contribute to AD pathogenesis through interactions with the gut microbiota. It has been well established that these factors are tightly linked to changes in the microbiome [99,100,101][74][75][76]. In addition, the host’s APOE alleles (especially APOE4), representing the strongest AD genetic risk factor, have been correlated with both an elevated innate immune response and changes in the butyrate-producing bacterial populations and their metabolite profiles in human and mouse models [102,103][77][78]. Similarly, several genetic risk genes for AD, including TREM2, CD33 (also known as SIGLEC-3), and SHIP1 have also been implicated in intestinal inflammation [104[79][80][81],105,106], which can directly or indirectly interact with the gut microbiota. The close relationship between gut dysbiosis, systemic inflammation, and AD pathogenesis has been shown in a clinical study where increases or decreases in the bacterial populations harboring either pro-inflammatory (e.g., Escherichia/Shigella) or anti-inflammatory (e.g., Eubacterium rectale) activities, respectively, were associated with systemic inflammation status and brain–amyloid deposition in cognitively impaired patients [107][82]. Similarly, a population study of IBD patients revealed that increased intestinal inflammation (likely coinciding with gut dysbiosis) was associated with both a higher early onset risk of AD and a higher overall incidence of AD [101][76]. Numerous correlational studies of gut dysbiosis in AD patients and in AD mouse models (e.g., APP/PS1, 5XFAD, and P301L transgenic mice) have extensively characterized changes in microbial populations, and these results have been summarized in recent reviews [59,108,109][34][83][84]. The importance of the gut microbiota as a contributing factor to AD has been functionally validated using GF and antibiotic-treated transgenic AD mouse models that exhibited reduced AD-like symptoms [110,111][85][86].
In the multifactorial etiology of LOAD, brain inflammation, proposed as one of the most crucial processes for its development [112][87], is known to be closely associated with microbiota and permeability changes in the gut, as described above. In neurological disorders, the GIT barrier has been found to be defective due to gut dysbiosis and altered microbial metabolites such as SCFAs and tryptophan metabolites [14], and the integrity of the BBB has also been shown to be compromised during the early stages of AD, irrespective of amyloid β and tau accumulations [113,114,115][88][89][90]. Neuroinflammation-related activated microglia and the increased expression of pro-inflammatory cytokines in the brain may both originate from such permeability defects in the GITs and brains of AD patients. Such defects may also allow the direct entry of pro-inflammatory bacteria such as oral pathogens (e.g., Porphyromonas gingivalis) and their endotoxic components/metabolites into the brain [116,117][91][92]. For example, infiltrated lipopolysaccharide (LPS), a representative microbe-associated molecular pattern molecule (MAMP), potentially originated from those pathogens, has been shown to promote Aβ production and aggregation as well as neuroinflammation [118,119][93][94] or to indirectly activate cells lining these barriers and the peripheral nervous system to elicit systemic inflammation and facilitate immune cell infiltration into the brain and inflammatory cytokines [120][95].
Interestingly, specific microbiota members in humans can also produce bacterial amyloid proteins, such as Curli produced from the csgA gene in E. coli. These bacterial amyloid proteins can prime amyloidosis and brain proteinopathies in neurodegenerative diseases through cross-seeding activity to form β-sheet structure-mediated toxic aggregates in a “prion-like” fashion via the autonomic nervous system, or by eliciting gut inflammation and releasing inflammatory mediators [121][96]. Although interactions between Curli and amyloid β of AD have yet to be tested directly, it has recently been demonstrated that Curli promoted the aggregation and pathology of α-synuclein, the protein responsible for Parkinson’s disease progression in vitro and in vivo [122][97].
References
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379.
- Hoban, A.E.; Stilling, R.M.; Ryan, F.J.; Shanahan, F.; Dinan, T.G.; Claesson, M.J.; Clarke, G.; Cryan, J.F. Regulation of prefrontal cortex myelination by the microbiota. Transl. Psychiatry 2016, 6.
- Erny, D.; Hrabe de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977.
- Sampson, T.R.; Mazmanian, S.K. Control of brain development, function, and behavior by the microbiome. Cell Host Microbe 2015, 17, 565–576.
- Morais, L.H.; Schreiber, H.L.t.; Mazmanian, S.K. The gut microbiota-brain axis in behaviour and brain disorders. Nat. Rev. Microbiol. 2020.
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.N.; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275.
- Diaz Heijtz, R.; Wang, S.; Anuar, F.; Qian, Y.; Bjorkholm, B.; Samuelsson, A.; Hibberd, M.L.; Forssberg, H.; Pettersson, S. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. USA 2011, 108, 3047–3052.
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809.
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: A therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 9–21.
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017, 60, 1–12.
- Hayes, C.L.; Dong, J.; Galipeau, H.J.; Jury, J.; McCarville, J.; Huang, X.; Wang, X.Y.; Naidoo, A.; Anbazhagan, A.N.; Libertucci, J.; et al. Commensal microbiota induces colonic barrier structure and functions that contribute to homeostasis. Sci. Rep. 2018, 8, 14184.
- Jakobsson, H.E.; Rodriguez-Pineiro, A.M.; Schutte, A.; Ermund, A.; Boysen, P.; Bemark, M.; Sommer, F.; Backhed, F.; Hansson, G.C.; Johansson, M.E. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep. 2015, 16, 164–177.
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal permeability--a new target for disease prevention and therapy. BMC Gastroenterol. 2014, 14, 189.
- Kelly, J.R.; Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G.; Hyland, N.P. Breaking down the barriers: The gut microbiome, intestinal permeability and stress-related psychiatric disorders. Front. Cell. Neurosci. 2015, 9, 392.
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Toth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6.
- Ploger, S.; Stumpff, F.; Penner, G.B.; Schulzke, J.D.; Gabel, G.; Martens, H.; Shen, Z.; Gunzel, D.; Aschenbach, J.R. Microbial butyrate and its role for barrier function in the gastrointestinal tract. Ann. N. Y. Acad. Sci. 2012, 1258, 52–59.
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055.
- Kaelberer, M.M.; Buchanan, K.L.; Klein, M.E.; Barth, B.B.; Montoya, M.M.; Shen, X.; Bohorquez, D.V. A gut-brain neural circuit for nutrient sensory transduction. Science 2018, 361.
- Fani Maleki, A.; Rivest, S. Innate Immune Cells: Monocytes, Monocyte-Derived Macrophages and Microglia as Therapeutic Targets for Alzheimer’s Disease and Multiple Sclerosis. Front. Cell Neurosci. 2019, 13, 355.
- Malm, T.; Koistinaho, M.; Muona, A.; Magga, J.; Koistinaho, J. The role and therapeutic potential of monocytic cells in Alzheimer’s disease. Glia 2010, 58, 889–900.
- Theriault, P.; ElAli, A.; Rivest, S. The dynamics of monocytes and microglia in Alzheimer’s disease. Alzheimers Res. Ther. 2015, 7, 41.
- Michaud, J.P.; Bellavance, M.A.; Prefontaine, P.; Rivest, S. Real-time in vivo imaging reveals the ability of monocytes to clear vascular amyloid beta. Cell Rep. 2013, 5, 646–653.
- Baik, S.H.; Cha, M.Y.; Hyun, Y.M.; Cho, H.; Hamza, B.; Kim, D.K.; Han, S.H.; Choi, H.; Kim, K.H.; Moon, M.; et al. Migration of neutrophils targeting amyloid plaques in Alzheimer’s disease mouse model. Neurobiol. Aging 2014, 35, 1286–1292.
- Zenaro, E.; Pietronigro, E.; Della Bianca, V.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880–886.
- Togo, T.; Akiyama, H.; Iseki, E.; Kondo, H.; Ikeda, K.; Kato, M.; Oda, T.; Tsuchiya, K.; Kosaka, K. Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J. Neuroimmunol. 2002, 124, 83–92.
- Gate, D.; Saligrama, N.; Leventhal, O.; Yang, A.C.; Unger, M.S.; Middeldorp, J.; Chen, K.; Lehallier, B.; Channappa, D.; De Los Santos, M.B.; et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 2020, 577, 399–404.
- Paouri, E.; Georgopoulos, S. Systemic and CNS Inflammation Crosstalk: Implications for Alzheimer’s Disease. Curr. Alzheimer Res. 2019, 16, 559–574.
- Peng, L.; Li, Z.R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 2009, 139, 1619–1625.
- Lobionda, S.; Sittipo, P.; Kwon, H.Y.; Lee, Y.K. The Role of Gut Microbiota in Intestinal Inflammation with Respect to Diet and Extrinsic Stressors. Microorganisms 2019, 7, 271.
- Wang, X.; Sun, G.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.; Chu, X.; Yang, J.; Wang, H. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803.
- Wu, S.C.; Cao, Z.S.; Chang, K.M.; Juang, J.L. Intestinal microbial dysbiosis aggravates the progression of Alzheimer’s disease in Drosophila. Nat. Commun. 2017, 8, 24.
- Sharon, G.; Garg, N.; Debelius, J.; Knight, R.; Dorrestein, P.C.; Mazmanian, S.K. Specialized metabolites from the microbiome in health and disease. Cell Metab. 2014, 20, 719–730.
- Dalile, B.; Van Oudenhove, L.; Vervliet, B.; Verbeke, K. The role of short-chain fatty acids in microbiota–gut–brain communication. Nat. Rev. Gastroenterol. Hepatol. 2019, 1, 461–478.
- Liu, S.; Gao, J.; Zhu, M.; Liu, K.; Zhang, H.L. Gut Microbiota and Dysbiosis in Alzheimer’s Disease: Implications for Pathogenesis and Treatment. Mol. Neurobiol. 2020, 57, 5026–5043.
- Casanova, M.F.; Frye, R.E.; Gillberg, C.; Casanova, E.L. Editorial: Comorbidity and Autism Spectrum Disorder. Front. Psychiatry 2020, 11.
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215.
- Yuen, R.K.; Merico, D.; Bookman, M.; Howe, J.L.; Thiruvahindrapuram, B.; Patel, R.V.; Whitney, J.; Deflaux, N.; Bingham, J.; Wang, Z.; et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 2017, 20, 602–611.
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444.
- Meltzer, A.; Van de Water, J. The Role of the Immune System in Autism Spectrum Disorder. Neuropsychopharmacology 2017, 42, 284–298.
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 155, 1451–1463.
- Yenkoyan, K.; Grigoryan, A.; Fereshetyan, K.; Yepremyan, D. Advances in understanding the pathophysiology of autism spectrum disorders. Behav. Brain Res. 2017, 331, 92–101.
- Hashem, S.; Nisar, S.; Bhat, A.A.; Yadav, S.K.; Azeem, M.W.; Bagga, P.; Fakhro, K.; Reddy, R.; Frenneaux, M.P.; Haris, M. Genetics of structural and functional brain changes in autism spectrum disorder. Transl. Psychiatry 2020, 10, 229.
- Gladysz, D.; Krzywdzinska, A.; Hozyasz, K.K. Immune Abnormalities in Autism Spectrum Disorder-Could They Hold Promise for Causative Treatment? Mol. Neurobiol. 2018, 55, 6387–6435.
- Siniscalco, D.; Schultz, S.; Brigida, A.L.; Antonucci, N. Inflammation and Neuro-Immune Dysregulations in Autism Spectrum Disorders. Pharmaceuticals 2018, 11, 56.
- Mayer, E.A.; Padua, D.; Tillisch, K. Altered brain-gut axis in autism: Comorbidity or causative mechanisms? Bioessays 2014, 36, 933–939.
- Samsam, M.; Ahangari, R.; Naser, S.A. Pathophysiology of autism spectrum disorders: Revisiting gastrointestinal involvement and immune imbalance. World J. Gastroenterol. 2014, 20, 9942–9951.
- Vuong, H.E.; Hsiao, E.Y. Emerging Roles for the Gut Microbiome in Autism Spectrum Disorder. Biol. Psychiatry 2017, 81, 411–423.
- Desbonnet, L.; Clarke, G.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. Microbiota is essential for social development in the mouse. Mol. Psychiatry 2014, 19, 146–148.
- Buffington, S.A.; Di Prisco, G.V.; Auchtung, T.A.; Ajami, N.J.; Petrosino, J.F.; Costa-Mattioli, M. Microbial Reconstitution Reverses Maternal Diet-Induced Social and Synaptic Deficits in Offspring. Cell 2016, 165, 1762–1775.
- Fattorusso, A.; Di Genova, L.; Dell’Isola, G.B.; Mencaroni, E.; Esposito, S. Autism Spectrum Disorders and the Gut Microbiota. Nutrients 2019, 11, 521.
- Xu, M.; Xu, X.; Li, J.; Li, F. Association Between Gut Microbiota and Autism Spectrum Disorder: A Systematic Review and Meta-Analysis. Front. Psychiatry 2019, 10, 473.
- Bolte, E.R. Autism and Clostridium tetani. Med. Hypotheses 1998, 51, 133–144.
- Sharon, G.; Cruz, N.J.; Kang, D.-W.; Gandal, M.J.; Wang, B.; Kim, Y.-M.; Zink, E.M.; Casey, C.P.; Taylor, B.C.; Lane, C.J. Human gut microbiota from autism spectrum disorder promote behavioral symptoms in mice. Cell 2019, 177, 1600–1618.
- Sandler, R.H.; Finegold, S.M.; Bolte, E.R.; Buchanan, C.P.; Maxwell, A.P.; Vaisanen, M.L.; Nelson, M.N.; Wexler, H.M. Short-term benefit from oral vancomycin treatment of regressive-onset autism. J. Child. Neurol. 2000, 15, 429–435.
- Li, Q.; Han, Y.; Dy, A.B.C.; Hagerman, R.J. The Gut Microbiota and Autism Spectrum Disorders. Front. Cell Neurosci. 2017, 11, 120.
- Rohr, M.W.; Narasimhulu, C.A.; Rudeski-Rohr, T.A.; Parthasarathy, S. Negative Effects of a High-Fat Diet on Intestinal Permeability: A Review. Adv. Nutr. 2020, 11, 77–91.
- Israelyan, N.; Margolis, K.G. Serotonin as a link between the gut-brain-microbiome axis in autism spectrum disorders. Pharmacol. Res. 2018, 132, 1–6.
- Suzuki, T.; Yoshinaga, N.; Tanabe, S. Interleukin-6 (IL-6) regulates claudin-2 expression and tight junction permeability in intestinal epithelium. J. Biol. Chem. 2011, 286, 31263–31271.
- De Angelis, M.; Piccolo, M.; Vannini, L.; Siragusa, S.; De Giacomo, A.; Serrazzanetti, D.I.; Cristofori, F.; Guerzoni, M.E.; Gobbetti, M.; Francavilla, R. Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS ONE 2013, 8, e76993.
- Fiorentino, M.; Sapone, A.; Senger, S.; Camhi, S.S.; Kadzielski, S.M.; Buie, T.M.; Kelly, D.L.; Cascella, N.; Fasano, A. Blood-brain barrier and intestinal epithelial barrier alterations in autism spectrum disorders. Mol. Autism. 2016, 7, 49.
- Matta, S.M.; Hill-Yardin, E.L.; Crack, P.J. The influence of neuroinflammation in Autism Spectrum Disorder. Brain Behav. Immun. 2019, 79, 75–90.
- Saurman, V.; Margolis, K.G.; Luna, R.A. Autism Spectrum Disorder as a Brain-Gut-Microbiome Axis Disorder. Dig. Dis. Sci. 2020, 65, 818–828.
- Sgritta, M.; Dooling, S.W.; Buffington, S.A.; Momin, E.N.; Francis, M.B.; Britton, R.A.; Costa-Mattioli, M. Mechanisms Underlying Microbial-Mediated Changes in Social Behavior in Mouse Models of Autism Spectrum Disorder. Neuron 2019, 101, 246–259.e6.
- Napoli, E.; Duenas, N.; Giulivi, C. Potential therapeutic use of the ketogenic diet in autism spectrum disorders. Front. Pediatrics 2014, 2, 69.
- Newell, C.; Bomhof, M.R.; Reimer, R.A.; Hittel, D.S.; Rho, J.M.; Shearer, J. Ketogenic diet modifies the gut microbiota in a murine model of autism spectrum disorder. Mol. Autism. 2016, 7, 37.
- Ristori, M.V.; Quagliariello, A.; Reddel, S.; Ianiro, G.; Vicari, S.; Gasbarrini, A.; Putignani, L. Autism, Gastrointestinal Symptoms and Modulation of Gut Microbiota by Nutritional Interventions. Nutrients 2019, 11, 2812.
- Kang, D.W.; Adams, J.B.; Gregory, A.C.; Borody, T.; Chittick, L.; Fasano, A.; Khoruts, A.; Geis, E.; Maldonado, J.; McDonough-Means, S.; et al. Microbiota Transfer Therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: An open-label study. Microbiome 2017, 5, 10.
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608.
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339.
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581.
- Hu, N.; Yu, J.T.; Tan, L.; Wang, Y.L.; Sun, L.; Tan, L. Nutrition and the risk of Alzheimer’s disease. Biomed. Res. Int. 2013, 2013.
- Razay, G.; Vreugdenhil, A.; Wilcock, G. The metabolic syndrome and Alzheimer disease. Arch. Neurol. 2007, 64, 93–96.
- Neuner, S.M.; Tcw, J.; Goate, A.M. Genetic architecture of Alzheimer’s disease. Neurobiol. Dis. 2020, 143.
- DeJong, E.N.; Surette, M.G.; Bowdish, D.M.E. The Gut Microbiota and Unhealthy Aging: Disentangling Cause from Consequence. Cell Host Microbe 2020, 28, 180–189.
- Dabke, K.; Hendrick, G.; Devkota, S. The gut microbiome and metabolic syndrome. J. Clin. Investig. 2019, 129, 4050–4057.
- Zhang, M.; Zhao, D.; Zhou, G.; Li, C. Dietary Pattern, Gut Microbiota, and Alzheimer’s Disease. J. Agric. Food Chem. 2020, 68, 12800–12809.
- Gale, S.C.; Gao, L.; Mikacenic, C.; Coyle, S.M.; Rafaels, N.; Murray Dudenkov, T.; Madenspacher, J.H.; Draper, D.W.; Ge, W.; Aloor, J.J.; et al. APOepsilon4 is associated with enhanced in vivo innate immune responses in human subjects. J. Allergy Clin. Immunol. 2014, 134, 127–134.
- Tran, T.T.T.; Corsini, S.; Kellingray, L.; Hegarty, C.; Le Gall, G.; Narbad, A.; Muller, M.; Tejera, N.; O’Toole, P.W.; Minihane, A.M.; et al. APOE genotype influences the gut microbiome structure and function in humans and mice: Relevance for Alzheimer’s disease pathophysiology. FASEB J. 2019, 33, 8221–8231.
- Correale, C.; Genua, M.; Vetrano, S.; Mazzini, E.; Martinoli, C.; Spinelli, A.; Arena, V.; Peyrin-Biroulet, L.; Caprioli, F.; Passini, N.; et al. Bacterial sensor triggering receptor expressed on myeloid cells-2 regulates the mucosal inflammatory response. Gastroenterology 2013, 144, 346–356.
- Fernandes, S.; Iyer, S.; Kerr, W.G. Role of SHIP1 in cancer and mucosal inflammation. Ann. N. Y. Acad. Sci. 2013, 1280, 6–10.
- Lubbers, J.; Rodriguez, E.; van Kooyk, Y. Modulation of Immune Tolerance via Siglec-Sialic Acid Interactions. Front. Immunol. 2018, 9, 2807.
- Cattaneo, A.; Cattane, N.; Galluzzi, S.; Provasi, S.; Lopizzo, N.; Festari, C.; Ferrari, C.; Guerra, U.P.; Paghera, B.; Muscio, C. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 2017, 49, 60–68.
- Kowalski, K.; Mulak, A. Brain-Gut-Microbiota Axis in Alzheimer’s Disease. J. Neurogastroenterol. Motil. 2019, 25, 48–60.
- Seo, D.O.; Holtzman, D.M. Gut Microbiota: From the Forgotten Organ to a Potential Key Player in the Pathology of Alzheimer’s Disease. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 1232–1241.
- Minter, M.R.; Zhang, C.; Leone, V.; Ringus, D.L.; Zhang, X.; Oyler-Castrillo, P.; Musch, M.W.; Liao, F.; Ward, J.F.; Holtzman, D.M.; et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 2016, 6, 30028.
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.; Frisoni, G.; Neher, J.; Fåk, F.; Jucker, M.; Lasser, T. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7.
- Krstic, D.; Knuesel, I. Deciphering the mechanism underlying late-onset Alzheimer disease. Nat. Rev. Neurol. 2013, 9, 25–34.
- Bowman, G.L.; Kaye, J.A.; Moore, M.; Waichunas, D.; Carlson, N.E.; Quinn, J.F. Blood-brain barrier impairment in Alzheimer disease: Stability and functional significance. Neurology 2007, 68, 1809–1814.
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302.
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276.
- Zhao, Y.; Cong, L.; Jaber, V.; Lukiw, W.J. Microbiome-Derived Lipopolysaccharide Enriched in the Perinuclear Region of Alzheimer’s Disease Brain. Front. Immunol. 2017, 8, 1064.
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5.
- Sheng, J.G.; Bora, S.H.; Xu, G.; Borchelt, D.R.; Price, D.L.; Koliatsos, V.E. Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol. Dis. 2003, 14, 133–145.
- Bulgart, H.R.; Neczypor, E.W.; Wold, L.E.; Mackos, A.R. Microbial involvement in Alzheimer disease development and progression. Mol. Neurodegener. 2020, 15, 42.
- Brown, G.C. The endotoxin hypothesis of neurodegeneration. J. Neuroinflammation 2019, 16, 180.
- Friedland, R.P.; Chapman, M.R. The role of microbial amyloid in neurodegeneration. PLoS Pathog. 2017, 13.
- Sampson, T.R.; Challis, C.; Jain, N.; Moiseyenko, A.; Ladinsky, M.S.; Shastri, G.G.; Thron, T.; Needham, B.D.; Horvath, I.; Debelius, J.W.; et al. A gut bacterial amyloid promotes alpha-synuclein aggregation and motor impairment in mice. Elife 2020, 9.