Anti-tumor photodynamic therapy (PDT) is a unique oxidative stress-based modality that has proven highly effective on a variety of solid malignancies. Endogenous nitric oxide (NO) has a critical role in the therapeutic outcome of this modality.
- photodynamic therapy
- singlet oxygen
- photooxidative stress nitric oxide
- inducible nitric oxide synthase
- S-nitrosation
- tumor cell resistance
- tumor cell aggressiveness
- bystander effects
- BET proteins
- BET inhibitors
1. Introduction: Photodynamic Therapy
Anti-tumor photodynamic therapy (PDT) was introduced about 45 years ago as a novel clinical approach for selectively eradicating a variety of malignant solid tumors via cytotoxic photochemistry [1,2,3,4][1][2][3][4]. Many of these tumors were refractory to conventional chemotherapy or radiotherapy. PDT is a minimally invasive modality which exhibits little, if any, off-target cytotoxicity. Classical PDT consists of three operating components: (i) a photosensitizing agent (PS), (ii) PS photoexcitation by non-ionizing radiation (typically in the far visible-to-near infrared wavelength range), and (iii) molecular oxygen [2,3,4][2][3][4]. All three components must be engaged concurrently to activate the photodynamic process, and light delivery via fiber optic networks makes PDT highly selective for an intended tumor target. In 1995, Photofrin®, a hematoporphyrin oligomer, became the first PS to receive Federal Drug Administration approval for clinical PDT, with esophageal malignancies being treated initially [2]. Since then, PDT with Photofrin® and other PSs has been used on numerous other malignancies, including prostate, breast, cervical, head-and-neck, and brain (gliomas) [3,4][3][4]. Most PSs are innocuous until photoactivated and, unlike many chemotherapeutic agents, have few (if any) negative effects on normal tissues. A common photodynamic reaction in PDT (Type II process) involves energy transfer from photoexcited triplet state PS to ground state O2 (Figure 1), giving singlet molecular oxygen (1O2), a cytotoxic short-lived reactive oxygen species (ROS). For some PSs, electron transfer to O2 may occur (Type I process), resulting in the formation of superoxide (O2−•) and other free radical ROS (Figure 1). Similarly to 1O2, the latter can kill tumor cells by oxidizing vital proteins and lipids and/or activating death signaling pathways [3,4,5][3][4][5]. In addition to preexisting PSs, which are administered directly in various carriers, pro-PSs have been developed which can be converted to active PSs in target tumors. One prominent example is 5-aminolevulinic acid (ALA), which is metabolized to protoporphyrin IX (PpIX) via the heme biosynthetic pathway [6,7][6][7]. To accommodate pro-growth/expansion activity, this pathway is typically more active in transformed cells than normal counterparts, thus accounting (at least in part) for greater PpIX accumulation in the former [6,7][6][7]. PpIX builds up initially in mitochondria and, upon photoactivation, induces photodamage therein, often leading to cell death via intrinsic apoptosis [8]. PDT with well-established PSs such as Photofrin®, benzoporphin derivative (BPD), silicon phthalocyanine (Pc4), and ALA-induced PpIX usually hits cytoplasmic targets (lysosomes, endoplasmic reticulum, mitochondria) [3,4][3][4]. In contrast, radiotherapy and chemotherapy usually affect nuclear targets, such as cisplatin, causing DNA cross-linking [9,10][9][10]. Since PDT acts mainly in the cytoplasm, this may explain its effectiveness as a follow-up or combined treatment with DNA-targeted chemo- or radiotherapy. It is well known that many tumors exhibit an intrinsic or acquired resistance to the latter modalities, and this is now evident for PDT as well. For example, a PDT challenge may upregulate antioxidant or anti-apoptotic proteins [11]. Moreover, some cancer cells can upregulate membrane transporter ABCG2, thereby reducing PDT efficacy by exporting ALA-induced PpIX and other PSs [12].
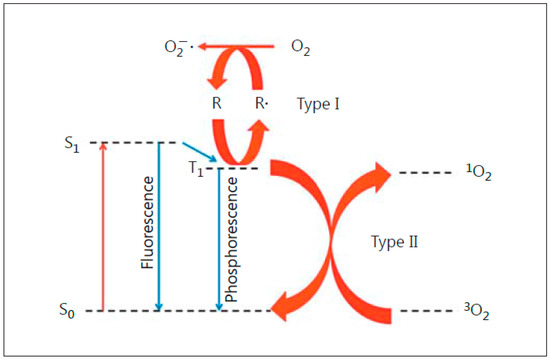
Figure 1. Photoexcitation of a sensitizing agent to a relatively long-lived triplet state (T1), which can initiate Type I (free radical-mediated) or Type II (singlet oxygen-mediated) reactions. Reproduced with permission from [4], Karger, 2015.
2. Role of Endogenous NO in Tumor Resistance to PDT
How endogenous NO might affect the anti-tumor potency of PDT at the in vivo level was first investigated about 20 years ago by two different groups using mouse syngeneic tumor models and Photofrin® as PS. Henderson et al. [34][13] found that PDT for mouse-borne RIF tumors was much improved when L-NNA, a competitive inhibitor of NOS activity, was administered before and after irradiation. At about the same time, Korbelik et al. [35,36][14][15] showed that the PDT cure rate for RIF and SCCVII tumors, but not EMT6 or FsaR tumors, could be significantly improved when L-NNA or L-NAME (another non-specific NOS inhibitor) was introduced immediately after irradiation. Importantly, RIF and SCCVII tumors were found to produce NO at a much higher constitutive rate than EMT6 or FsaR tumors, thus accounting for the greater inhibitor effects on the first two [36][15]. RIF and SCCVII tumors with relatively high NO outputs exhibited lower intrinsic sensitivity to PDT than the others, suggesting that basal NO measurements might serve as useful predictors of PDT efficacy [36][15]. More recently, Reeves et al. [37][16], also using mouse syngeneic tumors, but ALA-induced PpIX as PS, confirmed that tumor-generated NO exerted a negative effect on anti-tumor PDT. Essentially the same deduction was made in each of these studies, viz. that vasodilation due to endothelium-derived NO (presumably eNOS NO) was counteracting PDT’s well-known tumor-abating vasoconstrictive effects, allowing better maintenance of tumor oxygen supply during treatment. Although the earliest iteration of this work [34,35,36][13][14][15] was ground-breaking with regard to PDT antagonism by NO in vivo, it left the following questions unsettled: (i) whether the NO derives from tumor cells alone or whether surrounding cells (endothelial, macrophages, fibroblasts) or tumor-invading, PDT-induced cells like neutrophils might contribute; (ii) which NOS isoform is most important in any given tumor; (iii) whether the NOS/NO in question functions at a pre-existing level or is upregulated by PDT stress; and (iv) the biochemical mechanisms that underlie NO’s anti-PDT effects. Results of in vivo and in vitro studies dealing with these issues will be discussed in the following sections.
3. Role of NO in PDT-Induced Bystander Effects
As indicated above, most established tumors have defective vascular systems. Therefore, not all tumor cells would be uniformly supplied with an administered PS or pro-PS such as ALA. Moreover, during subsequent irradiation, some cells might only be exposed minimally or not at all due to insufficient light coverage, limited penetration, and scattering. Nevertheless, one could imagine that cells experiencing the greatest photodynamic stress might send signals to non- or minimally stressed neighboring cells (bystanders). Bystander effects are well documented for cancer cells exposed to ionizing radiation [62[17][18][19],63,64], and at least two means of signal transmission have been described: (i) via gap junctions between cells, and (ii) via the medium without actual cell contact [64][19]. Evidence for the latter has become more available as analytical methods have improved. Various signaling molecules capable of traversing aqueous media have been identified for X- or γ-irradiation, including cytokines [65][20], H2O2 [66][21], and NO [65,67,68,69][20][22][23][24]. Although NO has a short half-life and must be continuously generated to be effective, it differs from H2O2 in having no known enzymatic scavengers. PDT-induced bystander effects were first described about 20 years ago [70][25] and followed up 10 years later [71,72][26][27]. In the latter work, several signaling intermediates other than NO were proposed, e.g., H2O2, and LOOHs. The first studies to consider NO as a mediator of PDT bystander effects were carried out by Bazak et al. [73][28]. A novel silicone ring-based approach was used for initially separating ALA/light-targeted PC3 cells from non-targeted PC3 bystanders [73][28]. The rings were removed at some point after irradiation (typically 2–3 h), allowing any stress-upregulated factors to move from targeted cells to naïve bystanders. There was no physical contact between the two populations, so diffusion of signaling molecules through the medium was the only means of intercellular communication. As anticipated from earlier findings [56[29][30],57], photostress-withstanding target cells overexpressed iNOS, and its NO stimulated their proliferation and migration. Strikingly similar responses were observed in non-stressed bystanders, i.e., 1400W- and cPTIO-inhibitable iNOS/NO upregulation and faster proliferation and migration. Since the conditioned medium from targeted cells did not elicit these bystander responses [73][28], it was concluded that NO played the dominant role. Several other tumor-supporting proteins besides iNOS were upregulated in PC3 bystanders, including cyclooxygenase-2 (COX-2) and protein kinases Akt and ERK1/2. Once again, targeted cell iNOS/NO was primarily responsible [73][28]. These seminal studies on PC3 cells have recently been extended to three other human cancer lines: melanoma BLM, breast MDA-MB-231, and brain U87 [74][31]. Pre-incubation with ALA was the same for all these cells, but light fluences were varied, meaning that a uniform target cell kill was attained, i.e., ~25% at 24 h after irradiation. iNOS in both cell compartments underwent progressive upregulation over 24 h after irradiation, but to the following extents: PC3 > MDA-MB-231 > U87 > BLM. Proliferation and migration rates of targeted and bystander cells were enhanced in the same order. For example, BLM cells with the lowest iNOS induction exhibited the smallest increase in proliferation/migration rate, whereas PC3 cells with the greatest induction exhibited the greatest rate increase [74][31]. Thus, bystander aggressiveness increased in direct proportion to the extent of iNOS and NO upregulation in targeted cells that survived the ALA/light insult. A type of relay process is envisaged from these [74][31] and earlier findings [73][28], whereby NO overproduced in targeted cells diffuses to naïve bystanders and induces iNOS/NO there. How incoming NO does this in the bystander compartment, is still unknown. These findings suggest a NO “feed-forward” phenomenon that propagates through the bystander population. Such possibilities raise concerns about the tumor-promoting potential of bystander effects if they occur during clinical PDT.
(References would be added automatically after the entry is online)
References
- Dougherty, T.J.; Grindey, G.B.; Fiel, R.; Weishaupt, K.R.; Boyle, D.G. Photoradiation therapy II: Cure of animal tumors with hematoporphyrin and light. J. Natl. Cancer Inst. 1975, 55, 115–121.
- Dougherty, T.J.; Gomer, C.J.; Henderson, B.W.; Jori, G.; Kessel, D.; Korbelik, M.; Moan, J.; Peng, Q. Photodynamic therapy. J. Natl. Cancer Inst. 1998, 90, 889–905.
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281.
- Benov, L. Photodynamic therapy: Current status and future directions. Med. Pract. 2015, 24, 14–28.
- Girotti, A.W. Photosensitized oxidation of membrane lipids: Reaction pathways, cytotoxic effects, and cytoprotective mechanisms. J. Photochem. Photobiol. B 2001, 63, 103–113.
- Kennedy, J.C.; Pottier, R.H. Endogenous protoporphyrin IX, a clinically useful photosensitizer for photodynamic therapy. J. Photochem. Photobiol. B 1992, 14, 275–292.
- Peng, Q.; Berg, K.; Moan, J.; Kongshaug, M.; Nesland, J.M. 5-Aminolevulinic acid-based photodynamic therapy: Principles and experimental research. Photochem. Photobiol. 1997, 65, 235–251.
- Kim, R. Recent advances in understanding the cell death pathways activated by anticancer therapy. Cancer 2005, 103, 1551–1560.
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279.
- Hei, T.K.; Show, H.; Chai, Y.; Ponnaiya, B.; Ivanov, V.N. Radiation-induced non-targeted response: Mechanism and potential clinical implications. Curr. Mol. Pharmacol. 2011, 4, 96–105.
- Casas, A.; DiVenosa, G.; Hasan, T. Mechanisms of resistance to photodynamic therapy. Curr. Med. Chem. 2011, 18, 2486–2515.
- Palasuberniam, P.; Yang, X.; Kraus, D.; Jones, P.; Myers, K.A.; Chen, B. ABCG2 transporter inhibitor restores the sensitivity of triple negative breast cancer cells to aminolevulinic acid-mediated photodynamic therapy. Sci. Rep. 2015, 5, 13298.
- Henderson, B.W.; Sitnik-Busch, T.M.; Vaughn, L.A. Potentiation of photodynamic therapy antitumor activity in mice by nitric oxide synthase inhibition is fluence rate dependent. Photochem. Photobiol. 1999, 70, 64–71.
- Korbelik, M.; Shibuya, H.; Cecic, I. Relevance of nitric oxide to the response of tumors to photodynamic therapy. SPIE Proc. 1998, 3247, 98–105.
- Korbelik, M.; Parkins, C.S.; Shibuya, H.; Cecic, I.; Stratford, R.M.L.; Chaplin, D.J. Nitric oxide production by tumor tissue: Impact on the response to photodynamic therapy. Br. J. Cancer 2000, 82, 1835–1843.
- Reeves, K.L.; Reed, M.W.R.; Brown, N.J. The role of nitric oxide in the treatment of tumours with aminolaevulinic acid-induced photodynamic therapy. J. Photochem. Photobiol. B 2010, 101, 224–232.
- Azzam, E.I.; de Toledo, S.M.; Little, J.B. Stress signaling from irradiated to non-irradiated cells. Curr. Cancer Drug Targets 2006, 4, 53–64.
- Baskar, R. Emerging role of radiation-induced bystander effects: Cell communications and carcinogenesis. Genome Integr. 2010, 1, 13.
- Hei, T.K.; Zho, H.; Ivanov, V.N.; Hong, M.; Lieberman, H.B.; Brenner, d.J.; Amundson, S.A.; Geard, C.R. Mechanism of radiation-induced bystander effects: A unifying model. J. Pham. Pharmacol. 2008, 60, 943–950.
- Shao, C.; Folkard, M.; Prise, K.M. Role of TGF-beta1 and nitric oxide in the bystander response of irradiated glioma cells. Oncogene 2008, 27, 434–440.
- Pelle, E.; Mammone, T.; Maes, D.; Frenkel, K. Keratinocytes act as a source of reactive oxygen species by transferring hydrogen peroxide to melanocytes. J. Investig. Dermatol. 2005, 124, 793–797.
- Matsumoto, H.; Hayashi, S.; Hatashita, M.; Ohnishi, K.; Shioura, H.; Ohtsubo, T.; Kitai, R.; Ohnishi, T.; Kano, E. Induction of radioresistance by a nitric oxide-mediated bystander effect. Radiat. Res. 2001, 155, 387–396.
- Shao, C.; Stewart, V.; Folkard, M.; Michael, B.D.; Prise, K.M. Nitric oxide-mediated signaling in the bystander response of individually targeted glioma cells. Cancer Res. 2003, 63, 8437–8442.
- Yakovlev, V.A. Role of nitric oxide in the radiation-induced bystander effect. Redox Biol. 2015, 6, 396–400.
- Dahle, J.; Bagdonas, S.; Kaalhus, O.; Olsen, G.; Steen, H.B.; Moan, J. The by stander effect in photodynamic inactivation of cells. Biochim. Biophys. Acta 2000, 1475, 273–280.
- Chakraborty, A.; Held, K.D.; Prise, K.M.; Liber, H.L.; Redmond, R.W. Bystander effects induced by diffusing mediators after photodynamic stress. Radiat. Res. 2009, 172, 74–81.
- Rubio, N.; Rajadurai, A.; Held, K.D.; Prise, K.M.; Liber, H.L.; Redmond, R.W. Real-time imaging of novel spatial and temporal responses to photodynamic stress. Free Radic. Biol. Med. 2009, 47, 283–290.
- Bazak, J.; Fahey, J.M.; Wawak, K.; Korytowski, W.; Girotti, A.W. Enhanced aggressiveness of bystander cells in an anti-tumor photodynamic therapy model: Role of nitric oxide produced by targeted cells. Free Radic. Biol. Med. 2017, 102, 111–121.
- Bhowmick, R.; Girotti, A.W. Pro-survival and pro-growth effects of stress-induced nitric oxide in a prostate cancer photodynamic therapy model. Cancer Lett. 2014, 343, 115–122.
- Fahey, J.M.; Girotti, A.W. Accelerated migration and invasion of prostate cancer cells after a photodynamic therapy-like challenge: Role of nitric oxide. Nitric Oxide 2015, 49, 47–55.
- Bazak, J.; Korytowski, W.; Girotti, A.W. Bystander effects of nitric oxide in cellular models of anti-tumor photodynamic therapy. Cancers 2019, 11, 1674.