Despite significant recent improvements in the field of immunotherapy, cancer remains a heavy burden on patients and healthcare systems. In recent years, immunotherapies have led to remarkable strides in treating certain cancers. However, despite the success of checkpoint inhibitors and the advent of cellular therapies, novel strategies need to be explored to (1) improve treatment in patients where these approaches fail and (2) make such treatments widely and financially accessible. Vaccines based on tumor antigens (Ag) have emerged as an innovative strategy with the potential to address these areas. Here, we review the fundamental aspects relevant for the development of cancer vaccines and the critical role of dendritic cells (DCs) in this process. We first offer a general overview of DC biology and routes of Ag presentation eliciting effective T cell-mediated immune responses. We then present new therapeutic avenues specifically targeting Fc gamma receptors (FcγR) as a means to deliver antigen selectively to DCs and its effects on T-cell activation. We present an overview of the mechanistic aspects of FcγR-mediated DC targeting, as well as potential tumor vaccination strategies based on preclinical and translational studies. In particular, we highlight recent developments in the field of recombinant immune complex-like large molecules and their potential for DC-mediated tumor vaccination in the clinic. These findings go beyond cancer research and may be of relevance for other disease areas that could benefit from FcγR-targeted antigen delivery, such as autoimmunity and infectious diseases.
- Fc gamma receptors
- dendritic cells
- vaccination
- immune oncology
- recombinant immune complexes
1. Introduction
1.1. DCs Are Crucial for Effective Helper and Cytotoxic T-cell Activation
As suggested before, the effective activation of a T cell fully depends on its interaction with APCs [1][16]. They require an Ag to be presented in a rather short peptide sequence in a peptide:protein complex by the APC. The full activation of T cells requires the interplay of three different signals. Signal 1 is the recognition of the specific peptide presented to the T-cell receptor (TCR) by the major histocompatibility complex (MHC) molecules (either MHC-I for cytotoxic CD8+ T cells or MHC-II for CD4+ T cells) presented on APCs [2][3][17,18]. MHC-I-restricted peptides are mostly of cytoplasmic origin, while MHC-II-restricted peptides are of extracellular origin [3][18]. Through a process termed MHC-II cross presentation, extracellularly derived antigen peptides can be presented to CD8 T cells. This process itself is not sufficient to trigger the effective activation of Ag-specific T cells. In addition, they require Signal 2, characterized by the interaction between costimulatory molecules on T cells (e.g., CD28) and their counterparts on the APCs, such as CD80 and CD86 (also termed B7.1 and B7.2). Finally, to define the type of response, Signal 3 is required in the form of cytokines. Together, these three signals induce Ag-specific CD4+ or CD8+ T-cell responses [4][19]. Since DCs have the special ability to ingest virus-infected cells or tumor cells, they are able to present Ags derived from these to specific CD8+ T cells. The DCs activate them through a process termed cross-presentation via a separate MHC-I pathway [5][6][7][9,20,21].
DCs themselves become activated upon contact with foreign Ags [8][22]. DC activation can occur upon the engagement of conserved bacterial or viral Ags, so-called pathogen-associated molecular patterns (PAMPs) via pattern recognition receptors (PRRs). In resting conditions, immature DCs (imDCs) are equipped with several types of PRRs, including Toll-like receptors (TLRs), membrane-associated C-type lectin receptors (CLRs) [9][23], and mannose receptors [10][11][12][13][14][15][24,25,26,27,28,29]. Following the recognition of pathogens, imDCs can remain in a tolerogenic state [16][30] or undergo a maturation process where they lose their endocytic ability while increasing the Ag processing and presentation capacity [17][18][31,32]. PRR engagement activates mitogen-activated protein kinase (MAPK) and nuclear factor kappa-light-chain-enhancer of activated B cell (NF-κB) signaling [19][33], which, in mature DCs (maDCs), induces the expression of proinflammatory cytokines such as tumor necrosis factor alpha (TNF-α), interleukin-12 (IL-12), and IL-6 [20][34]. This is particularly important for the activation and clonal expansion of proinflammatory Th1-type CD4+ T cells [21][35]. MaDCs also upregulate chemokine receptors like CCR7 that drive their homing to lymph nodes (LN) [22][23][36,37]. The secretion of cytokines is reflected in a profound transcriptional change in DC gene expression that also results in the upregulation of Signal-2 markers, such as MHC-II, CD80, CD86, and CD40 [24][25][38,39]. Importantly, DC activation may alternatively trigger anti-inflammatory Th2 CD4+ T-cell activation or invigorate other specialized T-helper subsets, such as Th17, Th22, or regulatory T cells (Treg), depending on the context. We illustrate an overview of proinflammatory DC-mediated T-cell activation [26][40] in Figure 1.
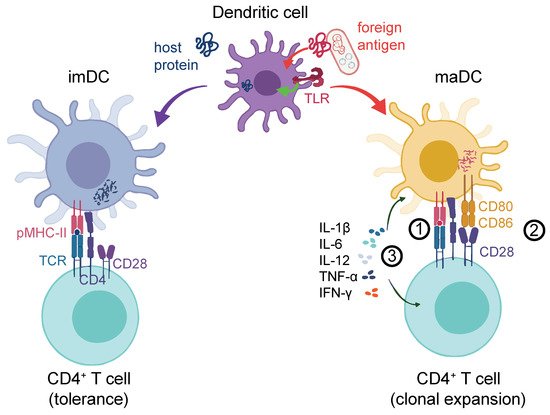
Figure 1. DC response to the antigen challenge. DCs can process either host-derived (self) proteins (blue, left-hand side) or foreign antigens (red, right-hand side). The latter could be from an exogeneous source (e.g., bacteria as illustrated) or cancer cell-derived neo-Ags. Self-protein processing and the presentation to T-cell receptors (signal 1) via peptide–MHC complexes (pMHC) leads to tolerance. In addition to signal 1, foreign antigens can lead to a strong DC activation, for instance, through the co-stimulation of TLRs or other receptors (not shown), which entails the upregulation of co-stimulatory molecules such as CD80 or CD86 at the DC surface (signal 2). These prolong and intensify the TCR-driven activation of antigen-specific T cells. Finally, cytokines such as IL-1β, IL-12, IL-6, IFN-γ, and TNF-α are released (signal 3) by both the DC and the T cell, which further shape the antigen-induced T-cell response. TCR: T cell receptor; pMHC-II: Peptide-MHC-II receptor; imDC: Immature DC; maDC: Mature DC.
PRRs are also relevant with regards to vaccine development, where the effective activation of T cells is critical. Although from a mechanistic point of view, targeting DCs seems like a promising avenue for vaccine development; it has been demonstrated that many DC vaccines alone do not achieve full T-cell activation [27][41]. In an effort to overcome this limitation, adjuvants can be used as key molecules aimed to promote stronger T-cell responses by inducing DC maturation and prolonging their exposure to antigens [27][41]. The efforts to create effective adjuvants have focused on the use of microbial compounds and selective TLR ligands [28][29][30][42,43,44]. However, PRRs are not only expressed on APCs but on a wide variety of myeloid cells, including neutrophils [31][45], monocytes [32][33][46,47], and macrophages [32][46], as well as nonimmune cells such as endothelial cells [34][48]. Consequently, the use of microbial compounds seemed to elicit a very broad inflammatory immune cell activation caused by non-DC PRR activation. Currently, even though adjuvants like TLR ligands [12][35][36][26,49,50], aluminum, or saponin-based particles are being studied to maximize the immunogenicity of vaccines [37][38][39][40][51,52,53,54], this strategy may still entail the risk of inducing general inflammation.
1.2. FcγR Crosslinking on DCs Leads to Effective T-Cell Activation and Proliferation
In addition to PRRs, DCs express Fc-gamma receptors (FcγR) that can lead to a highly effective internalization of Ag and subsequent DC activation [41][55]. FcγRs, when crosslinked through IgG antibody-complexed (“opsonized”) soluble Ag, allow for rapid internalization and cellular activation [42][43][56,57]. This immune complex (IC) will then be shuttled to endolysosomal DC compartments, where the Ag will subsequently be degraded. This facilitates the subsequent MHC:peptide generation and Ag presentation on the DC [42][44][56,58]. In humans, three groups of FcγRs have been described across a variety of cell types: FcγRI (CD64), FcγRIIA/B (CD32A/B), and IIIA/B (CD16A/B) [45][59]. Concerning DC activation, all FcγRs except FcγRIIB are considered activators; FcγRIIB acts as an inhibitory receptor. FcγRs bind—albeit, with different affinities [46][60]—to the Fc (fragment, crystallizable) portion of IgG antibodies [44][58]. On DCs, the expression of FcγRs depends on the cell subtype. FcγRI is expressed on monocyte-derived DCs (moDCs) [47][61]. FcγRIIA has been described on conventional DCs, which were also shown to express FcγRIIB [48][62]. In addition, human, as well as murine, pDCs were described to express FcγRII [49][50][51][52][63,64,65,66]. Overall, human and mouse DCs express largely overlapping FcγR subsets [53][54][67,68]. However, the balance between activating and inhibitory receptors on APCs critically depends on the tissue of origin, and the numbers of cell surface receptors can be different between the species [55][69].
Several lines of evidence, both in humans and mice, have convincingly demonstrated that IgG:Ag ICs induce a superior DC activation compared to the delivery of uncoated, “naked” Ag [42][43][56,57]. For example, pulsing in vitro human moDCs with polyclonal tetanus IgG ICs led to a stronger increase in DC activation, including the release of proinflammatory cytokines compared to “naked” tetanus toxoid Ag [43][56][57,70]. Similarly, in a pivotal mouse study, ovalbumin (OVA) preincubated with anti-OVA IgG was taken up much more efficiently by splenic mouse DCs ex vivo than “naked” OVA. Importantly, in mice transplanted with OVA-specific CD8+ or CD4+ T cells, OVA:IgG ICs induced both CD4+ and CD8+ T-cell proliferation more efficiently than “naked” OVA [57][71]. Similar experiments were repeated with henn egg lysozyme IgG ICs and with mice selectively lacking activating, as well as inactivating, FcγRs [41][57][58][55,71,72]. In addition, mechanistic confirmation was achieved using mouse models where signal transduction downstream of FcγRI and FcγRIIIA was impaired [41][59][55,73]. Another study showed that IC-mediated FcγR crosslinking in mouse DCs was required to induce long-lasting transcriptional changes reflected in the induction of T cell-polarizing genes, such as IL2, IL6, IL10, IL15, IL23a, IL27, and Ifnb1 [60][74]. These experiments provide the mechanistic basis to target FcγRs via IgG ICs, and this holds promise for DC-specific vaccination strategies [61][75].
2. Allogenic Tumor IgG ICs Can Trigger Cancer Immunity via DC Activation
Tumor rejection is thought to rely largely, if not completely, on the host’s effective immune response to tumor cells [62][63][76,77]. This process entails the immunosurveillance of potentially tumorigenic host cells by an intricate interplay between APCs and effector cells [64][78]. In cancer patients, DCs can present tissue-associated Ags or neo-Ags, which originate through cancer-specific DNA alterations [64][65][66][78,79,80]. This has led to the development of DC-selective tumor vaccination strategies [61][67][68][75,81,82].
In principle, both humoral, as well as cytotoxic T cell-mediated host immune responses, can lead to tumor rejection, depending on the tumor immunogenicity [69][70][83,84]. Interestingly, in mouse tumor systems such as the C57/BL6 B16F10 melanoma model [71][72][73][85,86,87], the rejection of tumors in allogenic recipient animals can be observed, suggesting pre-existing allogenic tumor immunity where immunocompetent mice reject allogenic (but not syngeneic) tumor cells post-transplantation. In a pivotal mouse study, Carmi et al. systematically assessed the mechanistic basis of allogenic tumor immunity and found that it was initiated by naturally occurring tumor-binding IgG, which enabled DCs to internalize tumor Ags and, subsequently, activate tumor-reactive T cells. Allogeneic tumors contained more maDCs than syngeneic tumors. The authors found that IgG binding to tumor cells was critical to initiate DC activation by using allogenic IgG fractions in conjunction with tumor cell lysates, thus generating tumor Ag:IgG ICs. Importantly, tumor Ag presentation following an antibody-mediated uptake by DCs was sufficient to initiate protective T cell-mediated immunity. This was confirmed in human cancer, where healthy donor IgG could form ICs with allogenic patient-derived lung carcinoma lysates. These and other results [57][74][75][76][71,88,89,90] prompted more mechanistic analyses of Ag:IgG IC-mediated cancer immunity.
3. FcγR-Targeted Vaccination Strategies in Preclinical Tumor Models
Mouse ex vivo cancer vaccination protocols involving the DC Ag challenge were developed as early as the 1990s [77][78][91,92]. As outlined before, the IgG IC:FcγR axis may provide an even more attractive angle for the design of DC-targeted strategies [79][80][93,94], leading to the development of IgG IC-mediated tumor vaccination models.
In an early congenic mouse melanoma model [81][95], the OVA-expressing B16F10 cell line MO-4 was used. Bone marrow-derived DCs (BMDCs) were generated from wildtype (wt) C57/BL6 animals and challenged in vitro with rabbit IgG:OVA ICs or “naked” OVA. IC:BMDC recipient animals were almost completely protected from tumor engraftment, while all control animals developed melanomas. This vaccination was much more efficient in inducing T-cell responses and longer-lasting compared to BMDCs challenged with “naked” OVA. In a recall experiment, OVA mice that had been vaccinated with the IgG IC BMDC protocol and subsequently survived MO-4 tumor cell transplantation were re-challenged with MO-4 a half-year later, and none of the animals developed palpable tumors. More importantly, from a therapeutic point of view, 40% of the tumor-bearing mice transplanted with OVA:IgG IC-challenged BMDCs could be rescued. This suggested that targeting DCs with IgG ICs might be exploited in tumor prevention, as well as in tumor treatment. Lastly, using C57Bl.6 β2M−/−, transporter associated with antigen-processing 1 (TAP1)−/−, MHC-II−/− and FcγRγ−/− animals, the authors confirmed that the vaccination depended on FcγRs and induced both MHC-I- and MHC-II-restricted responses.
Another mouse study by Schuurhuis et al. using OVA-expressing B16 tumor cells (here: MO-5) confirmed that the in vitro BMDC challenge with Ag:IgG IC s was superior to “naked” Ag stimulation [82][96]. Here, the differential contribution of mouse FcRγs was assessed by the selective and/or combined knockout (KO) of specific FcγRs. These comparisons showed that FcγRI and FcγRIII were required for enhancing the cross-presentation of CD8+ T cells, the critical effector T cells. In vitro, as well as in vivo, assays showed that FcγRI was found to compensate for the absence of FcγRIII and vice versa. Consequently, in this model, activating (but not inhibitory) FcγRs on BMDCs were required for the efficient priming of Ag-specific CD8+ T cells and induction of tumor protection. This confirmed again that, in tumor vaccination protocols, MHC-I−/− or MHC-II−/− DCs are unable to induce T cell-mediated tumor protection downstream of the DC Ag:IgG IC challenge [81][95]. Importantly, further experiments confirmed that transplanting BMDCs matured in vitro was more protective compared to mere OVA:IgG IC administration in a MO-5 melanoma induction model.
The Ag:IgG IC vaccination also induces functional humoral antibody responses to tumor Ags. In a recent study by Kim et al. [83][97], a recombinant Ag antibody IC was generated. Through the production of recombinant GA733, an epithelial cell adhesion molecule (EpCAM), Fc fusion protein, and an anti-GA733 mAb, anti-GA733 IgG ICs were obtained. These were subsequently administered to immunocompetent mice, leading to the induction of a Th2 response followed by the generation of (presumably polyclonal) anti-GA733 mouse antibodies. This antiserum was found to delay the growth of a human EpCAM+ colorectal cancer cell line in a nude mouse model. Upon tumor manifestation, serum derived from mock-challenged, GA733-challenged, or antibody:GA733 IC-challenged immunocompetent mice were transfused into recipients. Here, the antibody:GA733 IC-derived serum was found to be significantly superior at controlling tumor growth compared to GA733-challenged serum vaccination.
Taken together, IgG:Ag ICs provide an attractive entry route for therapeutic anticancer DC vaccination protocols with clear advantages over vaccinations with “naked” Ag. Importantly, the use of the whole Ag protein over human leukocyte antigen (HLA)-restricted peptide-based [84][98] tumor vaccination protocols could also mean more patients would be eligible for such treatments.
(References would be added automatically after the entry is online)