Iris integrity is required to regulate both the amount of light reaching the retina and intraocular pressure (IOP), with elevated IOP being a major risk factor for glaucoma. Congenital microcoria (MCOR) is an extremely rare, autosomal dominant disease affecting iris development and hindering both of these functions.
- congenital microcoria
- congenital miosis
- dilator muscle
- glaucoma
- myopia
- chromosome 13q32.1 structural variants
1. Introduction
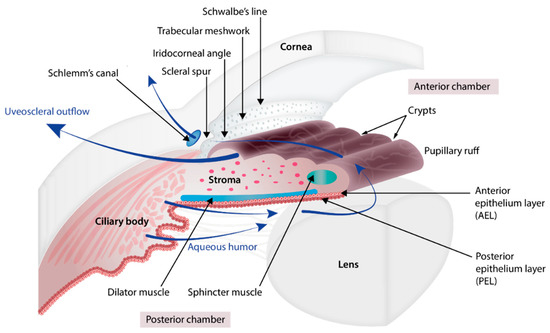
The iris (Figure 1 and Figure 2) is a flat ring-shaped ocular membrane located between the cornea and the lens. Its root is attached to the corneoscleral junction on the anterior side and the ciliary body on the posterior side, next to the lens, and its center is perforated to form the pupil. The iris is composed of a stroma, bilayer epithelium, and two smooth muscles that work in opposition to adapt the pupil aperture to light intensity [1]. The circular sphincter muscle lies within the stroma, near the pupil margin, and can constrict the pupil in miosis [2]. The dilator muscle extends longitudinally within the stroma, from the iris root to below the midpoint of the sphincter [3], and can contract to expand the pupillary aperture in mydriasis [1]. The dilator muscle originates from the anterior iris epithelium, composed of a single layer of myoepithelial cells, the basal portion of which consists in elongated smooth muscle processes [4]. The ciliary body is an extension of the iris, with which it is continuous. It produces a fluid known as aqueous humor that provides nourishment to eye structures. The aqueous humor flows between the iris and lens, through the pupil to the anterior part of the iris, where it is drained through the trabecular meshwork (TM), a sieve-like structure lying at the juncture of the corneoscleral region with the iris periphery [1]. Aqueous humor drainage is required to regulate IOP, which, when elevated, is a major risk factor for optic nerve damage (glaucoma) [5][6].
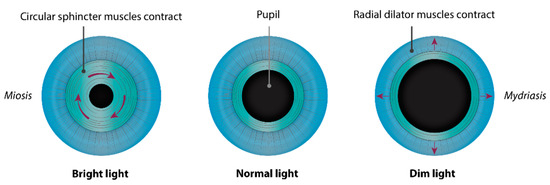
Congenital microcoria, also known as congenital miosis, is an iris malformation that affects both the regulation of the amount of light reaching the retina and the IOP. It is characterized by partial or total absence of dilator muscle fibers and manifested in pinhole pupils (<2 mm), iris hypopigmentation, and transillumination (Figure 2), causing both hemeralopia and light hypersensitivity [7][8][9][10][11][12]. Juvenile-onset glaucoma, axial myopia, and astigmatism are frequently associated with this condition, which can lead to visual dysfunction or blindness [8]. MCOR is a purely ocular disease. The presence of extraocular symptoms should evoke a differential diagnosis [13][14].
This is a very rare disease. Worldwide, some fifty families have been reported as having the disease, since its first mention about 150 years ago [15]. These cases describe autosomal dominant inheritance, and a unique locus has been mapped and ascribed to 13q32 submicroscopic chromosomal rearrangements [7].
2. Embryology of the Chamber Angle and Iris
The development of the eye involves the coordinated development of the neuroectoderm, surface ectoderm, and neural crest cell-derived mesenchyme.
The eye begins to develop as a pair of optic vesicles on each side of the forebrain around 3 weeks of gestation [16]. They extend from the forebrain toward the surface ectoderm through the adjacent mesenchyme. The contact of the optic vesicle with the surface ectoderm induces the thickening of the ectoderm, creating the lens placode which invaginates and detaches from the surface ectoderm to form the lens vesicle which will give rise to the lens. Mesenchyme cells begin to migrate into the space between the anterior epithelium of the lens vesicle and the surface ectoderm. The migration continues until the cells condense to form all the layers of the future cornea. The primitive corneal endothelial layer and future trabecular meshwork are formed from posterior mesenchyme cells whereas the surface ectoderm that covers the anterior side of the mesenchyme will become the corneal epithelium. In between, the mesenchyme cells differentiate to form the corneal stroma. During differentiation of the corneal endothelium, the lens detaches from the future cornea, creating the anterior chamber cavity [17].
Whilst the lens vesicle is forming, the optic vesicle also invaginates to form the double-walled optic cup. This iris and ciliary body derive from both the neuroectoderm and mesenchyme [18]. The epithelial layers of the iris and ciliary body, like the retina, develop from the third month of gestation as an outgrowth of the optic cup whilst the lens and the cornea are being formed. The outer wall produces pigment and forms the retinal pigment epithelium (RPE) and the inner wall differentiates to form the neural retina. The developing RPE and retina meet at the anterior rim of the optic cup, close to the lens vesicle which induces the differentiation of the cells of the inner wall of the anterior optic cup into the posterior pigmented epithelium of the iris (continuous with the developing retina) and the cells of the outer wall form the anterior iris epithelium (continuous with the developing RPE) [19]. At the root of the iris, the epithelium layers fold and the cells differentiate further to form the ciliary process epithelium [1]. The stroma of the iris and ciliary body arise from mesenchymal cells that migrate to the angle between the future cornea and the anterior edge of the optic cup which begins to extend to form the iris. The cells proliferate and migrate along the iris and ciliary body epithelial layers in formation and differentiate into stromal cells. Within the stroma of the iris, the sphincter pupillae and dilator pupillae muscles develop from optic cup neuroectodermal cells, contrasting with the ciliary muscle which derives from the mesenchyme [2]. The sphincter muscle arises from outer wall of the rim of the optic cup [20]. A group of cells characterized by diminished melanogenesis are distinguishable and indicate the future sphincter which begins to develop at 4 months and is well formed by 6 months [21]. The dilator pupillae also develops from the outer wall of the optic cup, but in a slightly more peripheral location than the sphincter. At the 6th month, myofilaments of the dilator muscles began to appear in the cytoplasm of the peripheral anterior pigment epithelium layer, with villous protrusions toward the stroma. The dilator muscle is fully formed histologically in human fetuses at eight months [22].
In parallel to the formation of the iris, the mesenchyme cells that migrated at the chamber angle separate from each other, generating small open spaces filled with extracellular fibers which will further organize into trabecular beams and vessels form close to the sclera which will ultimately form Schlemm’s canal [17]. Just posterior to the canal, tissue condenses to form the scleral spur that is composed of collagen and is continuous with that of the trabecular beams [5][23]. Between the trabecular beams and Schlemm’s canal, some mesenchymal cells differentiate into endothelial cells and fibroblasts which are embedded in a matrix of collagen, elastic-like fibers, and ground substance, forming the juxtacanalicular tissue [24]. While the TM is forming, the anterior chamber expands and its peripheral margin slides posteriorly, exposing the TM to the chamber cavity. The structures involved in aqueous humor drainage develop late, being mature around birth, and the anterior chamber is defined at 5 months of gestation [17].
3. Disease Description
The first mention of the disease dates back to 1862, when W. R. Wilde reported a series of three unrelated individuals displaying pinhole pupils without neurological problems, a condition he called “miosis congenita” [15]. In the following years, a dozen similar observations were reported by a few other ophthalmologists [25][26], some of whom described weakened pupil response to mydriatics [27]. To the best of our knowledge, a total of 160 affected individuals from 49 families have to date been reported with a bilateral disease characterized by partial or total absence of pupil dilation, even after mydriatic treatment [8][9]. A featureless surface with poorly developed collarette and crypts, reduced iris pigmentation, iris stroma thinning, and visible transillumination are typical of the disease [9][28][29][30][31][32].
Defective development of the iris musculature was suspected early on, as indicated by the phrases “fault of development” and “feeble development of the iris musculature” found in initial descriptions of the disease [16][17]. This was substantiated in 1923 by a Norwegian ophthalmologist and pathologist, who provided a detailed clinical description of the ocular phenotype in three siblings combined with a post mortem anatomical analysis of the eyes of two of them who had died of apoplexia cerebri [33]. This study, along with subsequent post mortem analyses of irises and iridectomy specimens from 25- to 72-year-old individuals, revealed significant iris thinning with atrophy of the stroma, displaying a normal ultrastructure and abundant collagen fibrils but greater numbers of fibroblasts and melanocytes in the ground substance [11][28][32][34][35][36][37]. Consistent with the observation of partial or total dilation inability among affected individuals, varying dilator muscle anomalies have also been described—from peripheral to generalized dearth or absence of stromal contractile processes of the anterior iris epithelium [8][9][10][11][21][34][35][38][39]. Existing myofibrils can be normal in appearance, particularly behind the sphincter muscle and in the intermediary region [33], or greatly disordered and lack myofilaments and desmin [9][28][33][34][35][37]. Thickened fibrobrotic and vacuolated dilator muscle can be observed [37]. The sphincter muscle and the ciliary body are normal, as are the innervation and vasculature [32][34][40]. In humans, the iris and ciliary body epithelia develop from the third month of gestation as an outgrowth of the anterior margins of the neuroectoderm-derived optic cup [41][42]. At 16 weeks of gestation, there is a distinct pigmented bilayer epithelium at the site that will later accommodate the adult iris. Myofilaments appear in the posterior iris epithelium near the presumptive pupillary margin in the 10th week, and in the cytoplasm of the peripheral anterior pigment epithelium in the 6th month, forming histologically recognizable sphincter and dilator muscles in the 6th and 8th months, respectively [43][44]. Differentiation of the anterior layer of the iris epithelium is manifested by the expression of α smooth muscle actin in the 28th week, and desmin intermediate filaments in dilator fibers by the 37th week [45]. Histological observations of all dilator muscle developmental stages, sometimes for the same iris [33]—ranging from a normal structure behind the sphincter, through poor differentiation with sparse and highly disordered fibers lacking myofilaments and intermediate filaments [9][10][37][46], down to complete growth inhibition [11][34]—suggest anomalies in the terminal stages of anterior iris pigment epithelium differentiation. Furthermore, this observation, which correlates with the variability of dilation phenotypes (partial or absent dilation ability), suggests that the genetic defect underlying the disease has a stochastic effect on development of the eye.
3.1. Associated Signs
3.1.1. Glaucoma
3.1.2. Axial Myopia
3.1.3. Astigmatism and Other Corneal Anomalies
3.1.4. Cataract
There have been 12 cases of late-onset or senile cataracts [11][21][28][67] and two cases of congenital cataracts [25][34] reported in individuals with congenital miosis. Their occurrence is likely coincidental considering that neither senile nor congenital cataracts are more prevalent in microcoric subjects than in the general population.
- Genetics
At least 24 multigenerational pedigrees (>100 cases in total) have been reported, while there have only been a dozen sporadic cases. There exist a moderate predominance of affected males (84 men, 63 females; M/F ratio: 1.3), but there is no difference in clinical presentation between the sexes. The disease is transmitted equally by males and females, and father-to-son transmission is not uncommon (at least 25 occurrences [10][29][61], demonstrating autosomal dominant inheritance. In multigenerational pedigrees, segregation analysis indicates the absence of disease transmission through unaffected obligate carriers, suggesting complete penetrance of the disease [8][9][10][29][30]. There is no evidence of genetic heterogeneity. Recent genetic studies have ascribed the disease to submicrosocopic structural variants of chromosome 13q32.1. Eight overlapping deletions ranging from 35.2 to 82 kilobases (kb) and one reciprocal 289 kb duplication have been reported in ten families from Europe (France, UK, Belgium, Switzerland), Middle East (Saudi-Arabia), Asia (Japan) and Central-America (Mexico) [7][12][31][32][67] (Figure 3). This observation suggests that the disease is due to a modification of the regulatory architecture of chromosome 13q32.1.
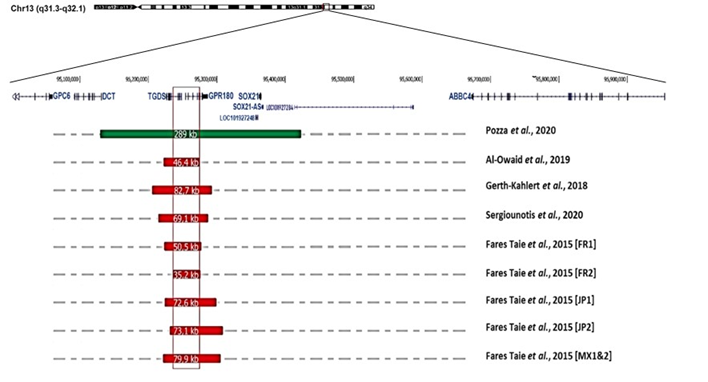
Figure 3. Gene organization at chromosome 13q32.1, and position and size of structural variations (deletions in red; duplication in green; according to the Human Genome Sequence Assembly hg19) reported in individuals suffering from congenital microcoria [7][12][31][32][67].
References
- Davis, N.; Ashery-Padan, R. Iris Development in Vertebrates; Genetic and Molecular Considerations. Brain Res. 2008, 1192, 17–28.
- Bloom, J.; Motlagh, M.; Czyz, C.N. Anatomy, Head and Neck, Eye Iris Sphincter Muscle. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021.
- Uvea. Clinical Gate 2015. Available online: (accessed on 8 March 2015).
- Forrester, J.; Dick, A.; McMenamin, P.; Roberts, F.; Pearlman, E. The Eye; Elsevier: Amsterdam, The Netherlands, 2016; ISBN1 978-0-7020-5554-6. Available online: (accessed on 19 February 2015)ISBN2 978-0-7020-5554-6.
- Abu-Hassan, D.W.; Acott, T.S.; Kelley, M.J. The Trabecular Meshwork: A Basic Review of Form and Function. J. Ocul. Biol. 2014, 2, 1–22.
- Goel, M.; Picciani, R.G.; Lee, R.K.; Bhattacharya, S.K. Aqueous Humor Dynamics: A Review. Open Ophthalmol. J. 2010, 4, 52–59.
- Fares-Taie, L.; Gerber, S.; Tawara, A.; Ramirez-Miranda, A.; Douet, J.-Y.; Verdin, H.; Guilloux, A.; Zenteno, J.C.; Kondo, H.; Moisset, H.; et al. Submicroscopic Deletions at 13q32.1 Cause Congenital Microcoria. Am. J. Hum. Genet. 2015, 96, 631–639.
- Toulemont, P.J.; Urvoy, M.; Coscas, G.; Lecallonnec, A.; Cuvilliers, A.F. Association of Congenital Microcoria with Myopia and Glaucoma. Ophthalmology 1995, 102, 193–198.
- Tawara, A.; Itou, K.; Kubota, T.; Harada, Y.; Tou, N.; Hirose, N. Congenital Microcoria Associated With Late-Onset Developmental Glaucoma. J. Glaucoma 2005, 14, 409–413.
- Rouillac, C.; Roche, O.; Marchant, D.; Bachner, L.; Kobetz, A.; Toulemont, P.-J.; Orssaud, C.; Urvoy, M.; Odent, S.; Le Marec, B.; et al. Mapping of a Congenital Microcoria Locus to 13q31-Q32. Am. J. Hum. Genet. 1998, 62, 1117–1122.
- Simpson, W.A.C. The Ultrastructural Pathological Features of Congenital Microcoria: A Case Report. Arch. Ophthalmol. 1989, 107, 99.
- Pozza, E.; Verdin, H.; Deconinck, H.; Dheedene, A.; Menten, B.; De Baere, E.; Balikova, I. Microcoria Due to First Duplication of 13q32.1 Including the GPR180 Gene and Maternal Mosaicism. Eur. J. Med. Genet 2020, 63, 103918.
- Pierson, M.; Cordier, J.; Hervouuet, F.; Rauber, G. An Unusual Congenital and Familial Congenital Malformative Combination Involving the Eye and KidneY. J. Genet Hum. 1963, 12, 184–213.
- Zenker, M.; Aigner, T.; Wendler, O.; Tralau, T.; Müntefering, H.; Fenski, R.; Pitz, S.; Schumacher, V.; Royer-Pokora, B.; Wühl, E.; et al. Human Laminin Β2 Deficiency Causes Congenital Nephrosis with Mesangial Sclerosis and Distinct Eye Abnormalities. Hum. Mol. Genet. 2004, 13, 2625–2632.
- Wilde, W.R. An Essay on the Malformations and Congenital Diseases of the Organs of Sight/by W. R. Wilde; John Churchill: London, UK, 1862.
- Edward, D.P.; Kaufman, L.M. Anatomy, Development, and Physiology of the Visual System. Pediatr. Clin. N. Am. 2003, 50, 1–23.
- Cvekl, A.; Tamm, E.R. Anterior Eye Development and Ocular Mesenchyme. Bioessays 2004, 26, 374–386.
- Bales, T.R.; Lopez, M.J.; Clark, J. Embryology, Eye. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021.
- Graw, J. Eye Development. Curr. Top. Dev. Biol. 2010, 90, 343–386.
- Ruprecht, K.W.; Wulle, K.G. Light and electron microscopic studies on the development of the human pupillary sphincter muscle. Albrecht Graefes Arch. Klin. Exp. Ophthalmol. Albrecht Graefe’s Arch. Clin. Exp. Ophthalmol. 1973, 186, 117–130.
- Polomeno, R.C.; Milot, J. Congenital Miosis. Can. J. Ophthalmol. 1979, 14, 43–46.
- Mann, I.C. Notes on the Anatomy of the Living Eye, as Revealed by the Gullstrand Slitlamp. J. Anat. 1925, 59, 155–165.
- Anderson, D.R. The Development of the Trabecular Meshwork and Its Abnormality in Primary Infantile Glaucoma. Trans. Am. Ophthalmol. Soc. 1981, 79, 458–485.
- Vranka, J.A.; Kelley, M.J.; Acott, T.S.; Keller, K.E. Extracellular Matrix in the Trabecular Meshwork: Intraocular Pressure Regulation and Dysregulation in Glaucoma. Exp. Eye Res. 2015, 133, 112–125.
- Treacher Collins, E.; Mayou, M.S. Pathology and Bacteriology in Ophthalmic Practice; William Heinemann: London, UK, 1911; pp. 64, 82.
- Wood, C.A. Fully Illustrated; Lenicet to Muscles, Ocular (Classic Reprint). In The American Encyclopedia and Dictionary of Ophthalmology; Forgotten Books: London, UK, 1917; Volume 10, ISBN 1396359575 (ISBN13: 9781396359576).
- Truc and Valude. Nouveaux Éléments d’Ophtalmologie; Maloine: Paris, France, 1896; p. 475.
- Ramirez-Miranda, A.; Paulin-Huerta, J.M.; Chavez-Mondragón, E.; Islas-de la Vega, G.; Rodriguez-Reyes, A. Ultrabiomicroscopic-Histopathologic Correlations in Individuals with Autosomal Dominant Congenital Microcoria: Three-Generation Family Report. Case Rep. Ophthalmol. 2011, 2, 160–165.
- Saxena, S.; Saxena, R.C. Congenital Microcoria: A Study in Three Generations. Indian J. Ophthalmol. 1993, 41, 130.
- Ramprasad, V.L.; Sripriya, S.; Ronnie, G.; Nancarrow, D.; Saxena, S.; Hemamalini, A.; Kumar, D.; Vijaya, L.; Kumaramanickavel, G. Genetic Homogeneity for Inherited Congenital Microcoria Loci in an Asian Indian Pedigree. Mol. Vis. 2005, 11, 934–940.
- Sergouniotis, P.I.; Ellingford, J.M.; O’Sullivan, J.; Fenerty, C.H.; Black, G.C. Genome Sequencing Identifies a Large Deletion at 13q32.1 as the Cause of Microcoria and Childhood-Onset Glaucoma. Acta Ophthalmol. 2017, 95, e249–e250.
- Gerth-Kahlert, C.; Maggi, J.; Töteberg-Harms, M.; Tiwari, A.; Budde, B.; Nürnberg, P.; Koller, S.; Berger, W. Absence of Goniodysgenesis in Patients with Chromosome 13Q Microdeletion-Related Microcoria. Ophthalmol. Glaucoma 2018, 1, 145–147.
- Holth, S.; Berner, O. Congenital miosis or pinhole pupils owing to developmental faults of the dilatator muscle. Br. J. Ophthalmol. 1923, 7, 401–419.
- Butler, J.M.; Raviola, G.; Miller, C.D.; Friedmann, A.I. Fine Structural Defects in a Case of Congenital Microcoria. Graefes Arch. Clin. Exp. Ophthalmol. 1989, 227, 88–94.
- Ferreira, B.F.D.A.; Schmidt, M.B.; Barbosa, L.J.; Oyamada, M.K.; Carricondo, P.C. Phacoemulsification and 1% Atropine Eye Drops for Treatment of Antimetropic Congenital Microcoria Associated with Cataracts. Arq. Bras. Oftalmol. 2019, 82.
- Lambert, S.R.; Amaya, L.; Taylor, D. Congenital Idiopathic Microcoria. Am. J. Ophthalmol. 1988, 106, 590–594.
- Pietropaolo, A.; Corvino, C.; DeBlasi, A.; Calabrò, F. Congenital Microcoria: Case Report and Histological Study. J. Pediatr. Ophthalmol. Strabismus. 1998, 35, 125–127.
- Bremner, F.D.; Houlden, H.; Smith, S.E. Genotypic and Phenotypic Heterogeneity in Familial Microcoria. Br. J. Ophthalmol. 2004, 88, 469–473.
- Meire, F.M.; Delleman, J.W. Autosomal Dominant Congenital Miosis with Megalocornea. Ophthalmic Paediatr. Genet. 1992, 13, 123–129.
- Hyams, S.W.; Neumann, E. Congenital Microcoria and Combined Mechanism Glaucoma. Am. J. Ophthalmol. 1969, 68, 326–327.
- Imaizumi, M.; Kuwabara, T. Development of the Rat Iris. Investig. Ophthalmol. 1971, 10, 733–744.
- Jensen, A.M. Potential Roles for BMP and Pax Genes in the Development of Iris Smooth Muscle. Dev. Dyn. 2005, 232, 385–392.
- Thumann, G. Development and Cellular Functions of the Iris Pigment Epithelium. Surv. Ophthalmol. 2001, 45, 345–354.
- Mann, I.C. The Development of the Human Iris. Br. J. Ophthalmol. 1925, 9, 495–512.
- Uusitalo, M.; Kivelä, T. Development of Cytoskeleton in Neuroectodermally Derived Epithelial and Muscle Cells of the Human Eye. Investig. Opthalmol. Vis. Sci. 1995, 36, 2584–2591.
- Mazzeo, V.; Gaiba, G.; Rossi, A. Hereditary Cases of Congenital Microcoria and Goniodysgenesis. Ophthalmic Paediatr. Genet. 1986, 7, 121–125.
- Veirs, E.R. Congenital Miosis: Associated with a Narrow Angle of the Anterior Chamber and Abnormally Placed Iris Tissue. Arch. Ophthalmol. 1961, 65, 59.
- Heatley, J. Miosis Congenita Familiar. Soc. Mex. Oftalmol. 1948, 22, 141–148.
- Ardouin, M.; Urvoy, M.; Lefranc, J. Microcorie Congenitale. Bull. Mem Soc. Fr. Ophtalmol. 1964, 77, 356.
- Stabilization of Glaucoma Associated with Microcoria|Semantic Scholar. Available online: (accessed on 9 March 2021).
- Sahori, A.; Katsumori, O.K. Congenital Miosis Associated with Juvenile Glaucoma. Folia Ophthalmol. Jpn. 1987, 38, 853–857.
- Coulon, G.; Delbosc, B.; Jeffredo, Y.; Viennet, G.; Oppermann, A.; Royer, J. Congenital microcoria: A case report with histopathological study. J. Fr. Ophtalmol. 1986, 9, 35–39.
- Hoskins, H.D.; Shaffer, R.N.; Hetherington, J. Anatomical Classification of the Developmental Glaucomas. Arch. Ophthalmol. 1984, 102, 1331–1336.
- Agoston, I.; Gróf, P. La Miose Congénitale et l’albinisme. OPH 1968, 155, 399–408.
- Acott, T.S.; Kelley, M.J. Extracellular Matrix in the Trabecular Meshwork. Exp. Eye Res. 2008, 86, 543–561.
- Mitchell, P.; Hourihan, F.; Sandbach, J.; Wang, J.J. The Relationship between Glaucoma and Myopia: The Blue Mountains Eye Study. Ophthalmology 1999, 106, 2010–2015.
- Quinn, G.E.; Berlin, J.A.; Young, T.L.; Ziylan, S.; Stone, R.A. Association of Intraocular Pressure and Myopia in Children. Ophthalmology 1995, 102, 180–185.
- Chen, S.-J.; Lu, P.; Zhang, W.-F.; Lu, J.-H. High Myopia as a Risk Factor in Primary Open Angle Glaucoma. Int. J. Ophthalmol. 2012, 5, 750–753.
- Sommer, A.; Tielsch, J.M. Risk Factors for Open-Angle Glaucoma: The Barbados Eye Study. JAMA Ophthalmol. 1996, 114.
- Schmid, K.L.; Hills, T.; Abbott, M.; Humphries, M.; Pyne, K.; Wildsoet, C.F. Relationship between Intraocular Pressure and Eye Growth in Chick. Ophthalmic Physiol. Opt. 2003, 23, 25–33.
- Hoyt, C.S.; Stone, R.D.; Fromer, C.; Billson, F.A. Monocular Axial Myopia Associated with Neonatal Eyelid Closure in Human Infants. Am. J. Ophthalmol. 1981, 91, 197–200.
- Rabin, J.; Sluyters, R.C.V.; Malach, R. Emmetropization: A Vision-Dependent Phenomenon. Investig. Opthalmol. Vis. Sci. 1981, 20, 561–564.
- Johnson, C.A.; Post, R.B.; Chalupa, L.M.; Lee, T.J. Monocular Deprivation in Humans: A Study of Identical Twins. Investig. Opthalmol. Vis. Sci. 1982, 23, 135–138.
- Raviola, E.; Wiesel, T.N. An Animal Model of Myopia. N. Engl. J. Med. 1985, 312, 1609–1615.
- Pardue, M.T.; Faulkner, A.E.; Fernandes, A.; Yin, H.; Schaeffel, F.; Williams, R.W.; Pozdeyev, N.; Iuvone, P.M. High Susceptibility to Experimental Myopia in a Mouse Model with a Retinal ON Pathway Defect. Investig. Opthalmol. Vis. Sci. 2008, 49, 706–712.
- Schaeffel, F.; Feldkaemper, M. Animal Models in Myopia Research. Clin. Exp. Opt. 2015, 98, 507–517.
- Al-Owaid, A.; Alarfaj, M.; Al-Qahtani, A.; Al-Arfaj, K. Congenital Microcoria in a Saudi Family. Ophthalmic Genet. 2019, 40, 578–580.
- Vail, D.T., Jr. Adult Hereditary Anterior Megalophthalmus Sine Glaucoma: A Definite Disease Entity: With Special Reference To The Extraction Of Cataract. Arch. Ophthalmol. 1931, 6, 39–62.