Adoptive cell therapy (ACT) with tumor-infiltrating T cells (TILs) has emerged as a promising therapy for the treatment of unresectable or metastatic solid tumors. One challenge to finding a universal anticancer treatment is the heterogeneity present between different tumors as a result of genetic instability associated with tumorigenesis. As the epitome of personalized medicine, TIL-ACT bypasses the issue of intertumoral heterogeneity by utilizing the patient’s existing antitumor immune response. Despite being one of the few therapies capable of inducing durable, complete tumor regression, many patients fail to respond. Recent research has focused on increasing therapeutic efficacy by refining various aspects of the TIL protocol, which includes the isolation, ex vivo expansion, and subsequent infusion of tumor specific lymphocytes.
- adoptive cell therapy
- tumor-infiltrating T cells
- immunotherapy
- metastatic treatment
1. Introduction
Solid malignancies encompass a wide range of diseases from lymphomas to carcinomas that affect nearly every organ in the body. The malignant transformation from normal cells is accompanied by progressive accumulation of genetic mutations. As a result, patients diagnosed with the same histological tumor type exhibit a wide range of genetic mutations and tumor microenvironments (TME). This inherent heterogeneity in tumors often leads to variations in patient-specific responses. Precision medicine, which refers to the tailoring of treatments to tumor-specific cellular or molecular characteristics, has recently became the mainstay of oncological therapy. The epitome of personalized therapy is adoptive cell transfer (ACT) with autologous tumor-infiltrating lymphocytes (TIL). This process involves tumor excision, isolation of TILs, ex vivo selection for autologous tumor reactivity, rapid expansion, and reinfusion of the T cell product back to the host [1] (Figure 1).
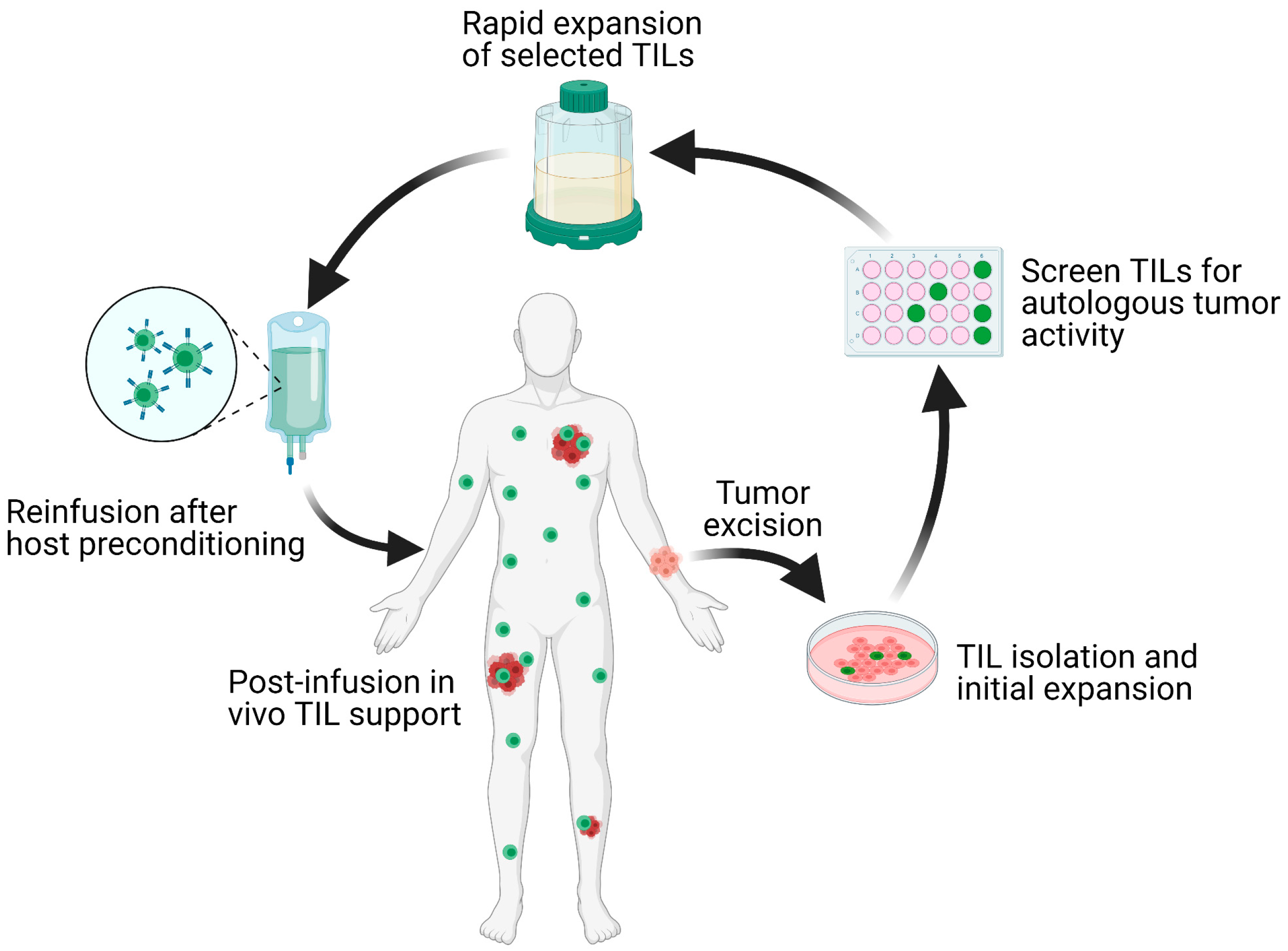
Figure 1.
10
The process of tumorigenesis produces nonsynonymous somatic mutations that lead to the generation of neoantigens. Neoantigens may be derived from (1) translation of mutated genes, (2) aberrant expression of non-mutated genes, and/or (3) the untimely expression of embryonic or cell-lineage specific genes [2][3]. In a similar stochastic process, every T cell expresses a unique T cell receptor (TCR) specific for one antigenic peptide sequence. Recognition of the cognate peptide presented on major histocompatibility complex (MHC) molecules lead to T cell activation and cytolytic target killing [4][5]. This diversity in potential neoantigens and endogenous TCRs poses a significant challenge for the effective immune recognition of tumors. As TIL-ACT specifically utilizes endogenous lymphocytes found within tumors, it increases the probability of enriching for tumor-specific T cells. Other ACT protocols have experimented with the transfer of different lymphocyte populations, including, but not limited to, genetically modified lymphocytes isolated from peripheral blood. In contrast to TILs, which contain endogenous tumor-recognition abilities, these lymphocytes are altered ex vivo to express tumor-specific TCRs and/or to have increased effector functions. [6]. A notable example, which is frequently used to treat hematological malignancies, is the transfer of genetically modified T cells with chimeric antigen receptors (CAR-T) that are specific for previously identified tumor-specific neoantigens [7].
The first evidence that ACT of autologous TILs leads to tumor-specific cytolysis and metastatic tumor control emerged in the 1980 s from murine models of sarcoma, colon adenocarcinoma, melanoma, and bladder carcinoma [8]. In the initial clinical study demonstrating the curative potential of TIL-ACT, 29 metastatic melanoma patients were treated with autologous TILs accompanied by high-dose interleukin-2 (IL-2), with a 31% objective response rate (ORR, complete response (CR) + partial response (PR)), and four patients achieving complete tumor regression [9]. A seminal clinical trial conducted by the Surgery Branch of the National Cancer Institute (NCI) in the early 2000 s demonstrated that the ORR for metastatic melanoma patients can be as high as 72%, with complete response rate (CRR) of 22% following one TIL infusion [10]. Therapeutic response to TIL-ACT is restricted by numerous factors, including, but not limited to, the initial amount of tumor-infiltrating T cells, the presence of immunosuppressive cell types in the TME, and the failure of adoptively transferred cells to persist in the host.
Advances in tumor immunology have led to the gradual optimization of the TIL-ACT protocol from lymphocyte selection to pre-treatment conditioning, and to combination with adjunctive treatment modalities.
2. Mechanisms of ACT-TIL Resistance
After the initial clinical trials in metastatic melanoma conducted by the NCI, TIL-ACT has been adopted by numerous other institutions around the world for the treatment of a variety of solid tumors [11][12][13][14]. While objective responses have been replicated in subsequent clinical trials, a significant percentage of patients experience therapy failure by displaying innate or acquired therapy resistance. Innate resistance occurs in patients unresponsive to initial therapy, whereas acquired resistance occurs in patients who were initially responsive but eventually become unresponsive. Regardless of when resistance occurs, the underlying mechanisms result in the lack of generation of functional T cells, or the suppression an otherwise productive T cell response (Figure 2).
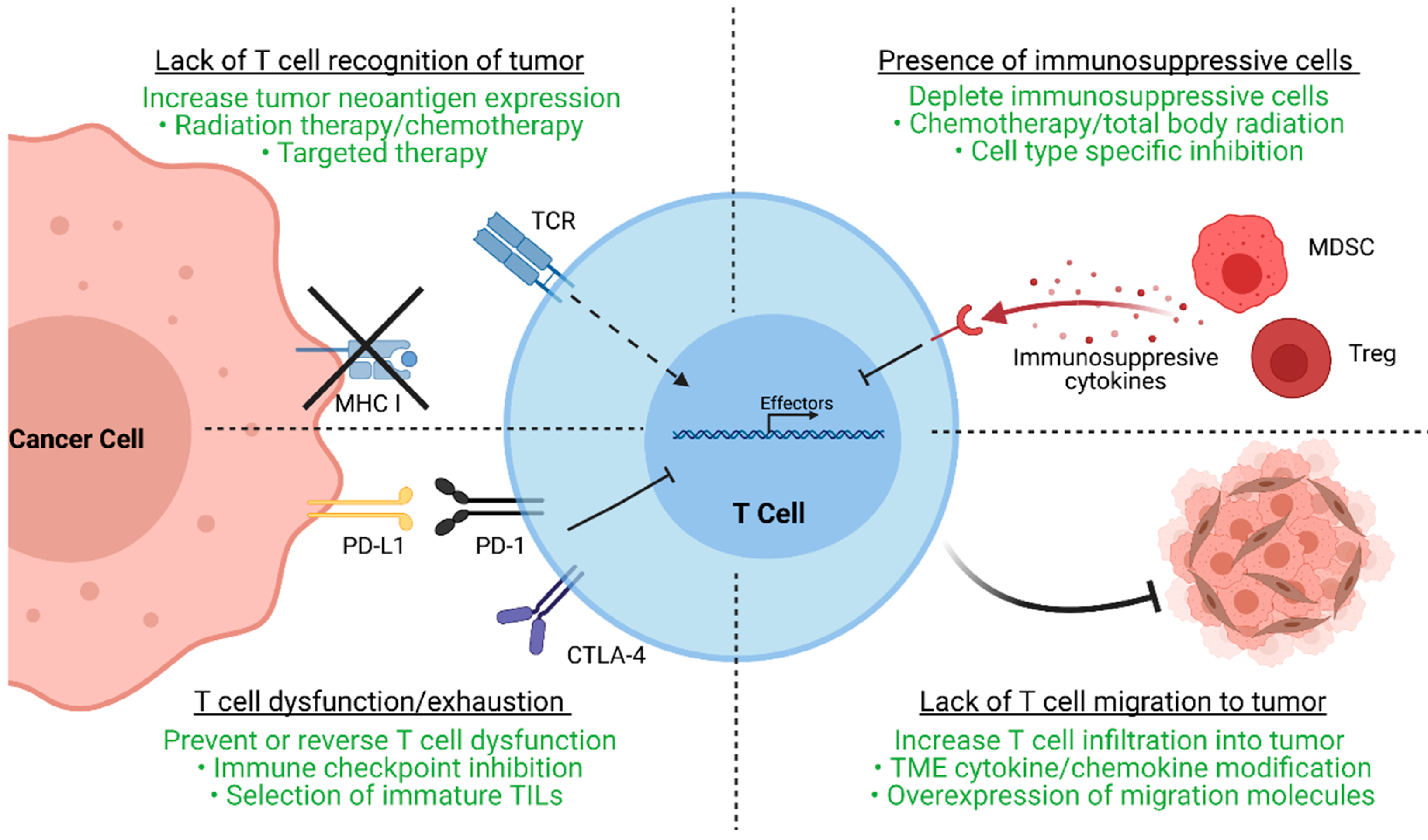
Figure 2.
As ACT is based on the premise of delivering antitumor T cells to the tumor, the lack of functional, tumor-specific T cells within tumor can abrogate therapeutic efficacy [12]. Effective TILs recognize tumor-specific neoantigens, which are results of stochastic somatic mutation accumulation during tumorigenesis. Thus, cancers with high mutational burdens, such as melanoma and lung cancer, have increased chances of generating neoantigens and are more likely to respond to T cell-based therapies [15][16][17]. In contrast, cancers with lower mutational burden, such as cervical cancer, exhibit decreased response to TIL- ACT (ORR 28%) [18]. However, cytolysis of neoantigen containing tumor clones may lead to the eventual outgrowth of antigen-negative tumor clones that are unrecognizable by TILs. Furthermore, tumor cells may also downregulate MHC molecules or other components of antigen presentation to evade immunosurveillance [19]. Aside from tumor cells, the TME can physically prevent TIL infiltration via generation of dense extracellular matrix, secretion of chemo-repulsive chemokines, or aberrant angiogenesis [20]. The TME can also contain a multitude of immunosuppressive cell types such as myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs). These cells can secrete anti-inflammatory cytokines (IL-10 and TGF-β), deplete essential T cell nutrients from the microenvironment, and suppress antigen presenting cells (APCs) to further inhibit T cell-mediated immunity [21][22]. Ultimately, different TME factors converge to generate a microenvironment conducive to T cell exhaustion, a state in which T cells upregulate checkpoint molecules as they progressively lose the ability to proliferative and perform effector functions [23]. In reality, numerous distinct escape mechanisms contribute to the immunosuppression and therapy resistance of TILs.
Given that every tumor is unique as a result of intra- and inter-tumoral heterogeneity, one must identify and target non-redundant resistance pathways present in each patient to optimize therapeutic benefit. With this knowledge, multiple clinical trials have focused on improving TIL-ACT responses by altering the original protocol based on four main components: (1) ex vivo selection and expansion of tumor-specific T lymphocytes; (2) patient pre-conditioning regimens; (3) post-infusion in vivo TIL support; and (4) potential adjuvant therapies.
3. The Future of TIL-ACT
Although the clinical success of TIL-ACT has been replicated across multiple institutions, its availability has so far been limited to large academic medical centers. These centers have the expertise to successfully grow TILs and the capability to adequately manage adverse events patients experience [24]. The ex vivo TIL expansion protocol is labor-intensive and the requirement to use fresh TIL products for infusion impose further restrictions on the implantation. Outsourcing TIL production to dedicated manufacturing centers is necessary to increase the cost-effectiveness and expand the availability of this therapy [25]. Iovance Biotherapeutics has established a centralized, high-throughput manufacturing facility for the production of cryopreserved TILs (brand named Lifleucel). Iovance’s model involve receiving surgically resected tumors shipped by different medical centers, expanding TILs to therapeutic numbers in their central facility, and shipping cryopreserved TIL products back to the medical centers [26] (Figure 3Figure 4). This alleviates the burdens on medical centers and theoretically allows TIL-ACT to be administered anywhere that has the capacity to surgically excise tumors and medically monitor patients during the reinfusion. Furthermore, cryopreserved TIL products offer more flexibility in scheduling as opposed to the rigid timeline dictated by the use fresh TILs. Despite the fact that TIL-ACT has yet to be FDA approved for the treatment of solid malignancies, strong preclinical and clinical results support its potential in eliciting durable tumor regression.
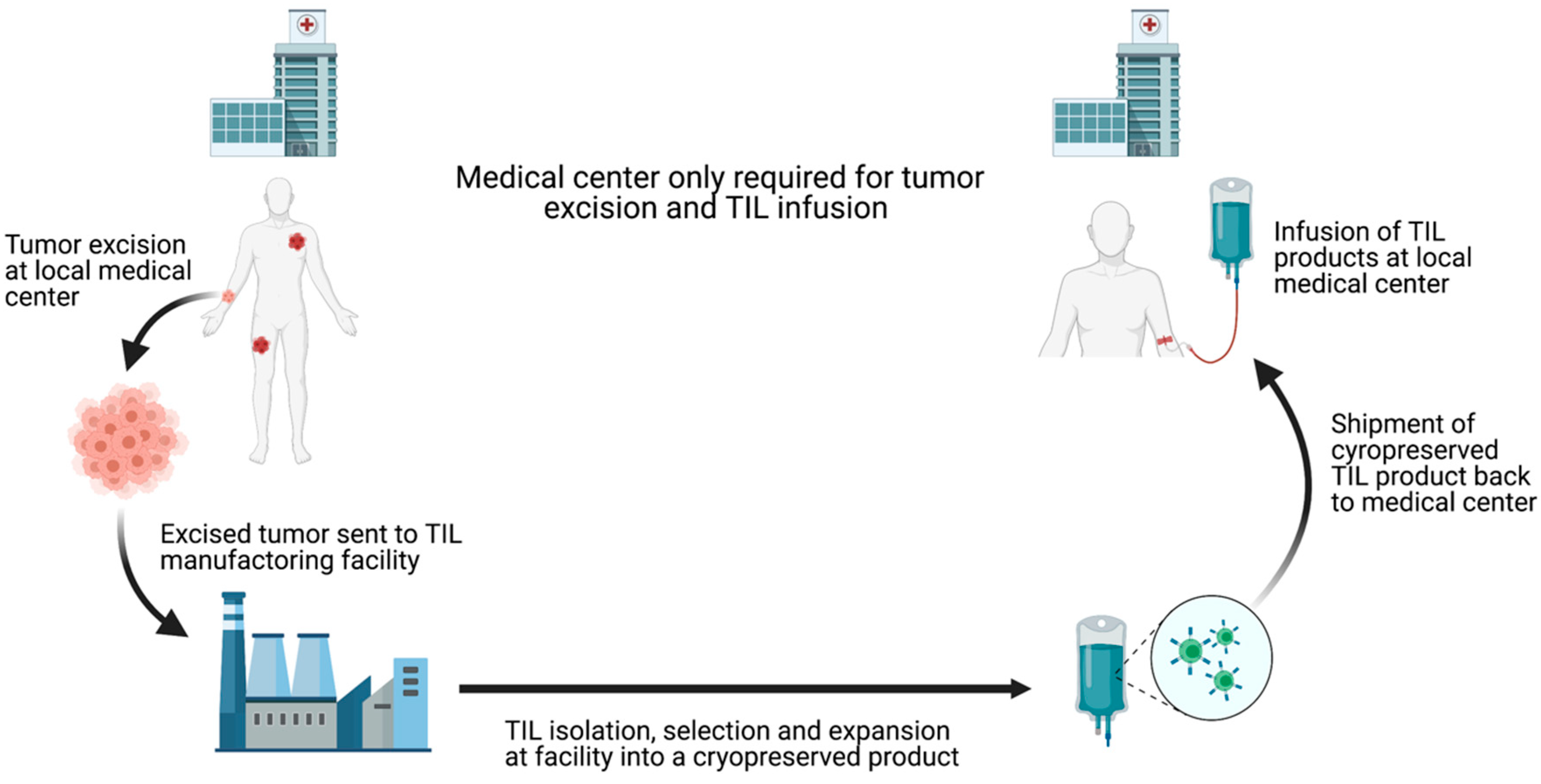
Figure 34. Schematic depiction of centralized TIL production chain. Commercialization of TIL production can increase TIL-ACT implantation by reducing the requirements for local medical centers. By outsourcing TIL production, treatment facilities only need to have the surgical and medical capacities to remove the tumor and treat potential infusion-associated adverse effects. TIL (tumor-infiltrating lymphocytes); ACT (adoptive cell therapy).
References
- Rohaan, M.W.; van den Berg, J.H.; Kvistborg, P.; Haanen, J. Adoptive transfer of tumor-infiltrating lymphocytes in melanoma: A viable treatment option. J. Immunother. Cancer 2018, 6, 102.
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200.
- Yarchoan, M.; Johnson, B.A., 3rd; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 569.
- Groscurth, P.; Filgueira, L. Killing Mechanisms of Cytotoxic T Lymphocytes. News Physiol. Sci 1998, 13, 17–21.
- Halle, S.; Halle, O.; Forster, R. Mechanisms and Dynamics of T Cell-Mediated Cytotoxicity In Vivo. Trends Immunol. 2017, 38, 432–443.
- Chandran, S.S.; Klebanoff, C.A. T cell receptor-based cancer immunotherapy: Emerging efficacy and pathways of resistance. Immunol. Rev. 2019, 290, 127–147.
- Rohaan, M.W.; Wilgenhof, S.; Haanen, J. Adoptive cellular therapies: The current landscape. Virchows Arch. 2019, 474, 449–461.
- Rosenberg, S.A.; Spiess, P.; Lafreniere, R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 1986, 233, 1318–1321.
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J. Natl. Cancer Inst. 1994, 86, 1159–1166.
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557.
- van den Berg, J.H.; Heemskerk, B.; van Rooij, N.; Gomez-Eerland, R.; Michels, S.; van Zon, M.; de Boer, R.; Bakker, N.A.M.; Jorritsma-Smit, A.; van Buuren, M.M.; et al. Tumor infiltrating lymphocytes (TIL) therapy in metastatic melanoma: Boosting of neoantigen-specific T cell reactivity and long-term follow-up. J. Immunother. Cancer 2020, 8, e000848.
- Pockaj, B.A.; Sherry, R.M.; Wei, J.P.; Yannelli, J.R.; Carter, C.S.; Leitman, S.F.; Carasquillo, J.A.; Steinberg, S.M.; Rosenberg, S.A.; Yang, J.C. Localization of 111indium-labeled tumor infiltrating lymphocytes to tumor in patients receiving adoptive immunotherapy. Augmentation with cyclophosphamide and correlation with response. Cancer 1994, 73, 1731–1737.
- Itzhaki, O.; Hovav, E.; Ziporen, Y.; Levy, D.; Kubi, A.; Zikich, D.; Hershkovitz, L.; Treves, A.J.; Shalmon, B.; Zippel, D.; et al. Establishment and large-scale expansion of minimally cultured “young” tumor infiltrating lymphocytes for adoptive transfer therapy. J. Immunother. 2011, 34, 212–220.
- Besser, M.J.; Shapira-Frommer, R.; Itzhaki, O.; Treves, A.J.; Zippel, D.B.; Levy, D.; Kubi, A.; Shoshani, N.; Zikich, D.; Ohayon, Y.; et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: Intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin. Cancer Res. 2013, 19, 4792–4800.
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. ‘Final common pathway’ of human cancer immunotherapy: Targeting random somatic mutations. Nat. Immunol. 2017, 18, 255–262.
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489.
- Alexandrov, L.B.; Stratton, M.R. Mutational signatures: The patterns of somatic mutations hidden in cancer genomes. Curr. Opin. Genet. Dev. 2014, 24, 52–60.
- Stevanovic, S.; Helman, S.R.; Wunderlich, J.R.; Langhan, M.M.; Doran, S.L.; Kwong, M.L.M.; Somerville, R.P.T.; Klebanoff, C.A.; Kammula, U.S.; Sherry, R.M.; et al. A Phase II Study of Tumor-infiltrating Lymphocyte Therapy for Human Papillomavirus-associated Epithelial Cancers. Clin. Cancer Res. 2019, 25, 1486–1493.
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692.
- van der Woude, L.L.; Gorris, M.A.J.; Halilovic, A.; Figdor, C.G.; de Vries, I.J.M. Migrating into the Tumor: A Roadmap for T Cells. Trends Cancer 2017, 3, 797–808.
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268.
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089.
- van der Leun, A.M.; Thommen, D.S.; Schumacher, T.N. CD8(+) T cell states in human cancer: Insights from single-cell analysis. Nat. Rev. Cancer 2020, 20, 218–232.
- Lindenberg, M.A.; Retel, V.P.; van den Berg, J.H.; Geukes Foppen, M.H.; Haanen, J.B.; van Harten, W.H. Treatment With Tumor-infiltrating Lymphocytes in Advanced Melanoma: Evaluation of Early Clinical Implementation of an Advanced Therapy Medicinal Product. J. Immunother. 2018, 41, 413–425.
- Lindenberg, M.; Retel, V.; Rohaan, M.; van den Berg, J.; Haanen, J.; van Harten, W. Evaluating different adoption scenarios for TIL-therapy and the influence on its (early) cost-effectiveness. BMC Cancer 2020, 20, 712.
- Fardis, M.; DiTrapani, K.; Chartier, C.; Finckenstein, F.G. Current and future directions for tumor infiltrating lymphocyte therapy for the treatment of solid tumors. Cell Gene Ther. Insights 2020, 6, 855–863.