VEGF-A (also called VEGF) is a member of the mammalian platelet-derived growth factor (PDGF) supergene family which also includes VEGF-B, VEGF-C, VEGF-D and placental growth factor (PlGF).
- kidney
- VEGF signaling
- toxicity
1. VEGF, Pro-Angiogenic Factor, Renal Expression
The most important molecule that promotes angiogenesis and increases vascular permeability is VEGF-A (also called VEGF). It is a member of the mammalian platelet-derived growth factor (PDGF) supergene family which also includes VEGF-B, VEGF-C, VEGF-D and placental growth factor (PlGF). VEGF signals mainly through the VEGF receptor 2 (VEGFR2) and also binds VEGFR1, both members of the VEGFR tyrosine kinase family (VEGFR1, VEGFR2, and VEGFR3, encoded by the genes
FLT1 (Fms Related Receptor Tyrosine Kinase 1), KDR (Kinase Insert Domain Receptor)
, and
FLT4 (Fms Related Receptor Tyrosine Kinase 4), respectively), which is expressed at elevated levels in endothelial cells [1][2]. VEGFR1 has a substantially higher affinity for VEGF than for VEGFR2 but mediates a much lower pro-angiogenic activity [3]. VEGFR3 is mainly restricted to lymphatic endothelial cells.
, respectively), which is expressed at elevated levels in endothelial cells [8,9]. VEGFR1 has a substantially higher affinity for VEGF than for VEGFR2 but mediates a much lower pro-angiogenic activity [10]. VEGFR3 is mainly restricted to lymphatic endothelial cells.
Receptor tyrosine kinases (RTKs) have a similar molecular structure, with a ligand-binding site in the extracellular domain, a transmembrane region, and a cytoplasmic region that contains the protein tyrosine kinase (TK) domain with an ATP-binding site. Growth factors bind their specific extracellular domain and activate RTKs by inducing receptor dimerization, which, in turn, activates their autophosphorylation, thus initiating a cascade of downstream signaling events [4]. They are key regulators of critical cellular processes such as proliferation and differentiation, cell survival, metabolism, and migration.
Receptor tyrosine kinases (RTKs) have a similar molecular structure, with a ligand-binding site in the extracellular domain, a transmembrane region, and a cytoplasmic region that contains the protein tyrosine kinase (TK) domain with an ATP-binding site. Growth factors bind their specific extracellular domain and activate RTKs by inducing receptor dimerization, which, in turn, activates their autophosphorylation, thus initiating a cascade of downstream signaling events [11]. They are key regulators of critical cellular processes such as proliferation and differentiation, cell survival, metabolism, and migration.
Several studies have documented the importance of VEGF signaling in maintaining glomerular integrity [5][6][7]. In the glomerulus, VEGF is expressed and secreted by podocytes, which are highly differentiated visceral epithelial cells with foot processes, located on the urinary side of the glomerular basement membrane. VEGFRs are expressed on the surface of both endothelial cells and podocytes, even if the latter remains controversial [8][9][10][11][12]. Indeed, the autocrine effect of VEGF on the podocyte has been discussed. Bertuccio and Veron et al. have established that VEGFR2 is expressed in adult mouse podocytes and glomerular endothelial cells [10][13]. Müller-Deil et al. have identified VEGFRs in murine and human podocyte cell cultures [11], while Wang et al. have shown the expression and distribution of VEGFR2 both in endothelial cells and in podocytes by immune electron microscopy and immunofluorescence in human patients [9]. However, when Eremina et al. induced a podocyte-specific deletion of VEGFR2 in mice, they did not observe any effect on either glomerular development or function. They failed to detect any expression of VEGFR2 in podocytes [8].
Several studies have documented the importance of VEGF signaling in maintaining glomerular integrity [12,13,14]. In the glomerulus, VEGF is expressed and secreted by podocytes, which are highly differentiated visceral epithelial cells with foot processes, located on the urinary side of the glomerular basement membrane. VEGFRs are expressed on the surface of both endothelial cells and podocytes, even if the latter remains controversial [15,16,17,18,19]. Indeed, the autocrine effect of VEGF on the podocyte has been discussed. Bertuccio and Veron et al. have established that VEGFR2 is expressed in adult mouse podocytes and glomerular endothelial cells [17,20]. Müller-Deil et al. have identified VEGFRs in murine and human podocyte cell cultures [18], while Wang et al. have shown the expression and distribution of VEGFR2 both in endothelial cells and in podocytes by immune electron microscopy and immunofluorescence in human patients [16]. However, when Eremina et al. induced a podocyte-specific deletion of VEGFR2 in mice, they did not observe any effect on either glomerular development or function. They failed to detect any expression of VEGFR2 in podocytes [15].
This configuration allows a VEGF-mediated epithelial–endothelial crosstalk and contributes to the functional glomerular filtration barrier through survival, proliferation, and/or differentiation of the adjacent fenestrated glomerular capillary endothelial cells [14]. VEGF is known to exert bidirectional effects on podocytes, depending on its expression level. In adult mice, chronic VEGF knockout induced thrombotic microangiopathy (TMA) [6]. The best documented example for a pathological role of VEGF inhibition in the kidney is pre-eclampsia. Pre-eclampsia is a hypertensive disorder peculiar to pregnancy (4–5% of pregnant women). This systemic syndrome appears to originate in the placenta and is characterized by widespread maternal endothelial dysfunction, the presence of new-onset hypertension, and proteinuria or other end-organ damage occurring after 20 weeks of gestation [15]. The pathogenesis of pre-eclampsia relies on placental ischemia, abnormal spiral artery remodeling, and oxidative stress, leading to increased systemic levels of the circulating soluble form of VEGFR1 (also known as sFlt1, soluble Fms-like tyrosine kinase-1), an antagonist of VEGF, and PlGF [16][17]. Indeed, excess levels of the anti-angiogenic factor sFlt1 are associated with decreased circulating levels of VEGF and PlGF, resulting in maternal endothelial dysfunction, glomerular endotheliosis, and proteinuria, which may progress to thrombotic microangiopathy [15][18][19]. By contrast, podocyte VEGF overexpression induces collapsing glomerulopathy [10][14]. Renal expression of VEGF and its receptors is upregulated in patients with diabetic nephropathy, which induces new vessel formation in the kidney, stimulates renal hypertrophy, and causes proteinuria in experimental models [20][21]. An in vitro study showed that VEGF promotes podocyte survival through an autocrine pathway involving VEGFR2, inducing podocin upregulation and its association with CD2AP (CD2-associated protein), an adaptor molecule regulating podocyte actin polymerization [22]. Moreover, VEGFR2 interacts with nephrin, an adhesion protein and key regulator of podocyte survival via Akt signaling. Indeed, VEGFR2 is rapidly phosphorylated in response to VEGF and recruits the Src kinase Fyn, which binds to nephrin and initiates a cascade of phosphorylation, leading to actin cytoskeleton polymerization and actin stress fiber formation [13][23].
This configuration allows a VEGF-mediated epithelial–endothelial crosstalk and contributes to the functional glomerular filtration barrier through survival, proliferation, and/or differentiation of the adjacent fenestrated glomerular capillary endothelial cells [21]. VEGF is known to exert bidirectional effects on podocytes, depending on its expression level. In adult mice, chronic VEGF knockout induced thrombotic microangiopathy (TMA) [13]. The best documented example for a pathological role of VEGF inhibition in the kidney is pre-eclampsia. Pre-eclampsia is a hypertensive disorder peculiar to pregnancy (4–5% of pregnant women). This systemic syndrome appears to originate in the placenta and is characterized by widespread maternal endothelial dysfunction, the presence of new-onset hypertension, and proteinuria or other end-organ damage occurring after 20 weeks of gestation [22]. The pathogenesis of pre-eclampsia relies on placental ischemia, abnormal spiral artery remodeling, and oxidative stress, leading to increased systemic levels of the circulating soluble form of VEGFR1 (also known as sFlt1, soluble Fms-like tyrosine kinase-1), an antagonist of VEGF, and PlGF [23,24]. Indeed, excess levels of the anti-angiogenic factor sFlt1 are associated with decreased circulating levels of VEGF and PlGF, resulting in maternal endothelial dysfunction, glomerular endotheliosis, and proteinuria, which may progress to thrombotic microangiopathy [22,25,26]. By contrast, podocyte VEGF overexpression induces collapsing glomerulopathy [17,21]. Renal expression of VEGF and its receptors is upregulated in patients with diabetic nephropathy, which induces new vessel formation in the kidney, stimulates renal hypertrophy, and causes proteinuria in experimental models [27,28]. An in vitro study showed that VEGF promotes podocyte survival through an autocrine pathway involving VEGFR2, inducing podocin upregulation and its association with CD2AP (CD2-associated protein), an adaptor molecule regulating podocyte actin polymerization [29]. Moreover, VEGFR2 interacts with nephrin, an adhesion protein and key regulator of podocyte survival via Akt signaling. Indeed, VEGFR2 is rapidly phosphorylated in response to VEGF and recruits the Src kinase Fyn, which binds to nephrin and initiates a cascade of phosphorylation, leading to actin cytoskeleton polymerization and actin stress fiber formation [20,30].
2. Anti-Angiogenic Drugs
Solid tumor growth and metastasis spreading depend on angiogenesis. Various signals may trigger the angiogenic switch, for example, metabolic stress (hypoxia, low pH, or hypoglycemia), mechanical stress, immune/inflammatory response, and genetic mutations [24][25][26]. VEGF secreted by tumor cells stimulates endothelial cell proliferation and survival, leading to the establishment of new blood vessels [3]. Indeed, hypoxia regulates angiogenesis at every step of this process through multiple pathways, including VEGF. The master oxygen homeostasis regulators of this process are the hypoxia-inducing factors, HIFs. The founding member of this family is HIF-1α [27].
Solid tumor growth and metastasis spreading depend on angiogenesis. Various signals may trigger the angiogenic switch, for example, metabolic stress (hypoxia, low pH, or hypoglycemia), mechanical stress, immune/inflammatory response, and genetic mutations [1,31,32]. VEGF secreted by tumor cells stimulates endothelial cell proliferation and survival, leading to the establishment of new blood vessels [10]. Indeed, hypoxia regulates angiogenesis at every step of this process through multiple pathways, including VEGF. The master oxygen homeostasis regulators of this process are the hypoxia-inducing factors, HIFs. The founding member of this family is HIF-1α [33].
In 1971, Folkman introduced anti-angiogenesis as a new anti-cancer strategy [28]. However, only in 2004, the US Food and Drug Administration (FDA) approved bevacizumab, a humanized anti-VEGF monoclonal antibody, for metastatic colorectal cancer treatment [29]. To date, several anti-angiogenic therapies have been developed and approved to treat cancers and other activated VEGFR-related diseases.
In 1971, Folkman introduced anti-angiogenesis as a new anti-cancer strategy [34]. However, only in 2004, the US Food and Drug Administration (FDA) approved bevacizumab, a humanized anti-VEGF monoclonal antibody, for metastatic colorectal cancer treatment [35]. To date, several anti-angiogenic therapies have been developed and approved to treat cancers and other activated VEGFR-related diseases.
2.1. Anti-VEGF mAb and Tyrosine Kinase Inhibitors
These drugs can be classified into two categories: small-molecule inhibitors that target the ATP-binding site of RTK intracellular domain (tyrosine kinase inhibitor, TKI), and monoclonal antibodies (mAbs) that either interfere with the RTK extracellular domain or target the VEGF ligand (anti-VEGF) [1][30] (
These drugs can be classified into two categories: small-molecule inhibitors that target the ATP-binding site of RTK intracellular domain (tyrosine kinase inhibitor, TKI), and monoclonal antibodies (mAbs) that either interfere with the RTK extracellular domain or target the VEGF ligand (anti-VEGF) [8,36] (
).
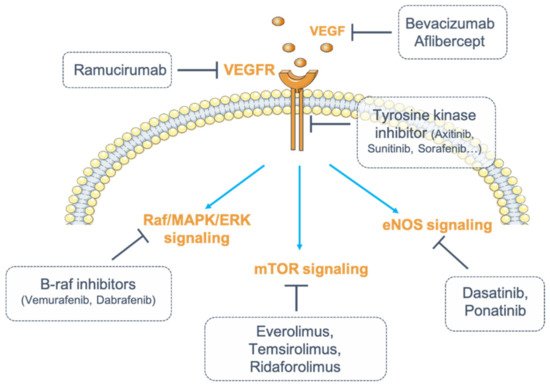
Figure 1. Inhibition of vascular endothelial growth factor (VEGF)/vascular endothelial growth factor receptor (VEGFR) signaling. Numerous strategies exist to inhibit VEGF/VEGFR signaling. VEGF can be blocked by monoclonal antibodies (mAbs) (bevacizumab) or by fusion proteins (aflibercept). Its receptor, VEGFR, can be targeted by fully humanized monoclonal antibodies (ramucirumab). The receptor can also be targeted for its intracellular tyrosine kinase activity (tyrosine kinase inhibitors, TKIs). Finally, one strategy consists of inhibiting the downstream signaling pathways of VEGFR by targeting either the Raf (Rapidly Accelerated Fibrosarcoma)/mitogen-activated protein kinase (MAPK)/ERK (Extracellular signal-Regulated Kinase) pathway with B-Raf inhibitors (dabrafenib, vemurafenib), or the endothelial nitric oxide synthase (eNOS) pathway (dasatinib, ponatinib) or the mammalian target of rapamycin (mTOR) pathway (everolimus, temsirolimus, ridaforolimus).
VEGF inhibitors through antibody binding include bevacizumab, ranibizumab, aflibercept, and ramucirumab. Bevacizumab and ranibizumab are monoclonal antibodies (mAbs), and aflibercept is a recombinant fusion protein that acts as a soluble decoy receptor or VEGF trap. Ramucirumab is a fully humanized mAb that specifically inhibits VEGFR2. Among these angiogenesis inhibitors, some are used either alone or in combination with chemotherapy, while most TKIs are multi-kinase inhibitors targeting VEGFRs and other RTKs simultaneously [31] (
VEGF inhibitors through antibody binding include bevacizumab, ranibizumab, aflibercept, and ramucirumab. Bevacizumab and ranibizumab are monoclonal antibodies (mAbs), and aflibercept is a recombinant fusion protein that acts as a soluble decoy receptor or VEGF trap. Ramucirumab is a fully humanized mAb that specifically inhibits VEGFR2. Among these angiogenesis inhibitors, some are used either alone or in combination with chemotherapy, while most TKIs are multi-kinase inhibitors targeting VEGFRs and other RTKs simultaneously [37] (
).
Table 1.
Incidence of renal manifestations and electrolytic disorders under anti-angiogenic targeted therapies.
| Drugs | Molecular Targets | Tumor Targets | Adverse Events (Incidence) | Electrolytic Disorders | References |
|---|---|---|---|---|---|
| Monoclonal antibodies | |||||
| Bevacizumab | VEGF | CRC, NSCLC, RCC, GBM, epithelial ovarian cancer, primary peritoneal cancer, cervical cancer, fallopian cancer, glioblastoma, ocular diseases | HTN (23–41%), Proteinuria (2–32%) | Hypophosphatemia, Hyponatremia | [32][33]39[34][35][36][37][38][39][40],40[,4141,42][,4342][38,,44,45,46,47,48] |
| Ranibizumab | VEGF | Ocular diseases | HTN, Proteinuria | - | [32][33][38,39] |
| Ramucirumab | VEGFR2 | CRC, NSCLC, GC | HTN, Proteinuria | - | [32][33][38,39] |
| Recombinant fusion protein | |||||
| Aflibercept | VEGF | CRC, ocular diseases | HTN, Proteinuria | - | [32][33][38,39] |
| Multitargeted TKI | |||||
| Sorafenib | VEGFRs, PDGFRs, RAF, c-Kit, FLT3, Ret | RCC, HCC, DTC | HTN (17–55%), Proteinuria (10%) | Hypophosphataemia (16–85%), Hyponatremia (39%) | [43][44][45][46][47][48][49[52][49,50][,5150][,5251,53],54,55,56,57,58] |
| Sunitinib | VEGFRs, PDGFRs, FLT3, CSF1R, Ret | RCC, GIST, pNETs | HTN (22–60%), Proteinuria (10–65%) | Hypophosphatemia, Hyponatremia | [53][54][55][56][57]][59,60[,6158],62[59],63[,6460,65,66] |
| Pazopanib | VEGFRs, PDGFRs, FGFR1, c-Kit | RCC, STS | HTN (40–52%), Proteinuria (13.5–18%) | Hypophosphatemia (34%), Hypocalcemia (33%), Hyponatremia (31%), Hypomagnesemia (11%) | [43][44][61][62][63][,5064,67],68,69[,7065][66][67][49,71,72,73] |
| Vandetanib | VEGFRs, EGFR, Ret | MTC | HTN (23.5–84%), Proteinuria (5.6–26%) | Hypomagnesemia (10–40%)Hypocalcemia (4–29%)Hypokaliemia (4–17%) | [68][44][69][70][71][72][73][74][5,50,74,75,76,77,78,79] |
| Axitinib | VEGFRs, PDGFRs, c-Kit | RCC | HTN (40–64%), Proteinuria (4.6–23%) | Hyponatremia, Hypophosphatemia (13%), Hypocalcemia (39%) | [68][43][44][75][76][77][78][5,49,50,80,81,82,83] |
| Regorafenib | VEGFRs, PDGFRs, FGFRs, Tie2, c-Kit, Ret, RAF | GIST, CRC, HCC | HTN (13–59%), Proteinuria (7–9.4%) | Hypophosphataemia (5–18%) | [68][43][44][49][79][80][81][5,49,50,55,84,85,86] |
| Cabozantib | VEGFRs, c-Met, AXL, c-Kit, FLT3, Ret | MTC, RCC | HTN (7–16), Proteinuria (6%) | Hypophosphatemia (4–8%) | [68][82][83][84][85][5[,8786,88,89],90,91] |
| Nintedanib | VEGFRs, PDGFRs, FGFRs, SRC | IPF, NSCLC | HTN, Proteinuria | - | [87][92] |
| Lenvatinib | VEGFRs, FGFRs, PDGFRa, Ret, c-Kit | DTC, RCC, HCC | HTN (45–100%), Proteinuria (26.9–100%) | Hypophosphatemia (45%) | [68][49][88][5,55,93] |
| Dasatinib | BCR-ABL, SRC, LCK, YES, FYN, c-Kit, VEGFR, PDGFR | CML, Ph+ ALL | Proteinuria | Hyponatremia | [89][94[90],95] |
| Ponatinib | VEGFRs, BCR-ABL, FLT3, Ret, c-Kit, FGFRs, PDGFR | CML, Ph+ ALL | HTN (9–32%) | - | [91][92][93][96,97,98] |
CML: chronic myeloid leukemia; CRC: colorectal cancer; CSF1R: colony stimulating factor 1 receptor; DTC: differentiated thyroid cancer; FGFR: fibroblast growth factor receptor; FLT3: fms like tyrosine kinase 3; GBM: glioblastoma multiforme; GC: gastric cancer (or gastroesophageal junction adenocarcinoma); GIST: gastrointestinal stromal tumor; HCC: hepatocellular carcinoma; HTN: hypertension; IPF: idiopathic pulmonary fibrosis; LCK: lymphocyte-specific protein tyrosine kinase; MTC: medullary thyroid cancer; NSCLC: non-small cell lung cancer; PDGFR: platelet-derived growth factor; Ph+ ALL: Philadelphia-chromosome-positive acute lymphoblastic leukemia; pNETs: progressive pancreatic neuroendocrine tumors; RAF: rapidly accelerated fibrosarcoma; RCC: renal cell carcinoma; STS: soft tissue sarcoma; VEGF: vascular endothelial growth factor; VEGFR: vascular endothelial growth factor receptor.