Autophagy is a catabolic process that ensures homeostasis in the cells of our organism. It plays a crucial role in protecting eye cells against oxidative damage and external stress factors. Ocular pathologies of high incidence, such as age-related macular degeneration, cataracts, glaucoma, and diabetic retinopathy are of multifactorial origin and are associated with genetic, environmental factors, age, and oxidative stress, among others; the latter factor is one of the most influential in ocular diseases, directly affecting the processes of autophagy activity.
- autophagy
- ocular pathology
- glaucoma
1. Introduction
Autophagy, a catabolic process that is conserved in the evolutionary process, involves the degradation and recycling of cellular components to maintain the cells’ interior under appropriate conditions for their survival [1]. The process of autophagy is a double-edged sword, as it is involved in both cell death and cell survival [1,2][1][2]. Through digestion, autophagy can eliminate debris that may be toxic, causing cell death. It can also digest cellular organelles, which are altered for subsequent recycling [3]. Autophagy also removes defective or aggregated proteins and lipid droplets from the cytoplasm via double membrane vesicles that are transported to and fused with lysosomes for enzymatic degradation [4]. After the digestion process, metabolites that can serve as the basis for the synthesis of new organelles or essential molecules for the cell are generated.
Various stress conditions, such as infections, ionizing radiation, hypoxia, and even chemotherapeutics, can cause the induction of autophagy [5]. Yoshinori Oshumi won the Nobel Prize in 2016 for his research work on autophagy processes. In the early 1990s, he identified genes related to autophagy processes in yeast (Saccharomyces cerevisiae) [6], subsequently characterizing the proteins encoded by these genes. He later conducted similar research in mammals [7]. Since the beginning of the 21st century, this catabolic process has been one of the main targets in cellular research in order to study some types of cancer, metabolic, inflammatory, and neurodegenerative diseases as well as cellular aging [1]. In addition, it is known that alterations in autophagy processes are involved in the development of certain ocular pathologies; therefore, treatments focused on the regulation of autophagy processes could offer a therapeutic alternative for the treatment of ophthalmologic pathologies [8].
32. Autophagy in Ocular Diseases
Under normal conditions, autophagy plays an important role in the maintenance of cellular homeostasis through the recycling of cytoplasmic proteins and cellular organelles [2]. However, disruption of normal autophagy processes is implicated in the pathophysiology of major ocular diseases [9][10][11][12].
2.1. Glaucoma
Oxidative stress plays a crucial role in the development of glaucoma by damaging the cells of [46the trabecular meshwork (TM), which are responsible for regulating the outflow of aqueous humor and maintaining intraocular pressure (IOP) [13]. Therefore, as an individual ages, oxidative damage accumulates, contributing to cell death by apoptosis and the development of glaucoma [14]. To protect themselves from constant oxidative stress and maintain intracellular homeostasis, TM cells activate autophagy to eliminate damaged proteins and organelles. Autophagic activity begins to saturate during the lifetime of an individual, since non-degradable materials accumulate in the lysosome compartment, which consequently causes a decrease in lysosomal activity. This decrease in autophagy leads to progressive dysfunction of TM cells and may contribute to the development of a glaucomatous pathology [15][16][17][18]. Elevated IOP can induce autophagy independently of mTOR to maintain cellular homeostasis in TM cells and cope with the mechanical forces by which they are affected. Porter et al. (2014) [16] conducted an in vitro experiment with pig eyes in which they applied a normal physiological pressure (8 mmHg) to some eyes (control eyes) and a high pressure to others (30 mmHg). Compared with controls, eyes subjected to high pressure showed increased levels of LC3-II (Microtubule-associated protein-1 light chain 3-Type II),47 a membrane component that is converted from LC3-I (Type I) to initiate autophagosome formation and elongation. In addition,48 ultrastructural analysis showed the presence of autophagosomes and autolysosomes in TM cells in those eyes subjected to high pressure,49] a result that was not observed in control eyes.
3.1. Glaucoma
Oxidative stress plays a crucial role in the development of glaucoma by damaging the cells of the trabecular meshwork (TM), which are responsible for regulating the outflow of aqueous humor and maintaining intraocular pressure (IOP) [50]. Therefore, as an individual ages, oxidative damage accumulates, contributing to cell death by apoptosis and the development of glaucoma [51]. To protect themselves from constant oxidative stress and maintain intracellular homeostasis, TM cells activate autophagy to eliminate damaged proteins and organelles. Autophagic activity begins to saturate during the lifetime of an individual, since non-degradable materials accumulate in the lysosome compartment, which consequently causes a decrease in lysosomal activity. This decrease in autophagy leads to progressive dysfunction of TM cells and may contribute to the development of a glaucomatous pathology [52,53,54,55]. Elevated IOP can induce autophagy independently of mTOR to maintain cellular homeostasis in TM cells and cope with the mechanical forces by which they are affected. Porter et al. (2014) [53] conducted an in vitro experiment with pig eyes in which they applied a normal physiological pressure (8 mmHg) to some eyes (control eyes) and a high pressure to others (30 mmHg). Compared with controls, eyes subjected to high pressure showed increased levels of LC3-II (Microtubule-associated protein-1 light chain 3-Type II), a membrane component that is converted from LC3-I (Type I) to initiate autophagosome formation and elongation. In addition, ultrastructural analysis showed the presence of autophagosomes and autolysosomes in TM cells in those eyes subjected to high pressure, a result that was not observed in control eyes.
In cultures of human TM cells derived from glaucomatous eyes in which IOP had been elevated for a long time, when cells were exposed to high oxygen concentrations (40% O
In cultures of human TM cells derived from glaucomatous eyes in which IOP had been elevated for a long time, when cells were exposed to high oxygen concentrations (40% O
2), autophagy processes were not activated [55]. Control TM cells from the healthy eye showed an autophagic response to hyperoxia, as there was an upregulation of LC3-II levels, but cells from glaucomatous eyes did not show a variation in this structural protein. Increased LC3 immunoreactivity in the RGC layer as well as LC3-II accumulation was also observed between 6 and 24 h after the transient increase in IOP in an experimental model developed in rats [70,71]. Impairment of the autophagy process by calpain-mediated cleavage of beclin-1 was also demonstrated in this same experimental model [72]. In a transgenic mouse model with altered basal autophagic flux by ablation of the Beclin-1-regulated autophagy-activated molecule (AMBRA-1), increased RGC loss in response to ischemia was observed after a transient increase in IOP [73]. Furthermore, in a transgenic mouse model with impaired autophagic flux and chronic hypertension DBA/2J::GFP-LC3, which express the autophagosome LC3, higher IOP values, further RGC loss, and aggravated axonal impairment were observed compared with the spontaneous ocular hypertensive DBA/2J mouse model [74]. All of these studies demonstrate that autophagy is rapidly activated at the onset of the disease in response to elevated IOP, but if these conditions were chronic autophagic activity would become saturated and cease to function properly. Therefore, the disruption of autophagic flux could be involved in the pathogenesis and progression of glaucoma [75] (
), autophagy processes were not activated [18]. Control TM cells from the healthy eye showed an autophagic response to hyperoxia, as there was an upregulation of LC3-II levels, but cells from glaucomatous eyes did not show a variation in this structural protein. Increased LC3 immunoreactivity in the RGC layer as well as LC3-II accumulation was also observed between 6 and 24 h after the transient increase in IOP in an experimental model developed in rats [19][20]. Impairment of the autophagy process by calpain-mediated cleavage of beclin-1 was also demonstrated in this same experimental model [21]. In a transgenic mouse model with altered basal autophagic flux by ablation of the Beclin-1-regulated autophagy-activated molecule (AMBRA-1), increased RGC loss in response to ischemia was observed after a transient increase in IOP [22]. Furthermore, in a transgenic mouse model with impaired autophagic flux and chronic hypertension DBA/2J::GFP-LC3, which express the autophagosome LC3, higher IOP values, further RGC loss, and aggravated axonal impairment were observed compared with the spontaneous ocular hypertensive DBA/2J mouse model [23]. All of these studies demonstrate that autophagy is rapidly activated at the onset of the disease in response to elevated IOP, but if these conditions were chronic autophagic activity would become saturated and cease to function properly. Therefore, the disruption of autophagic flux could be involved in the pathogenesis and progression of glaucoma [24] (
Figure 3).
1).
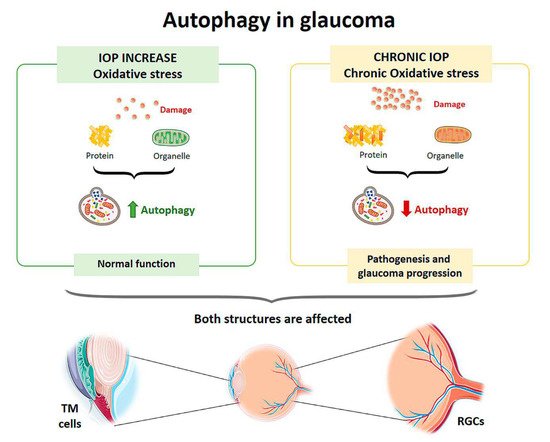
Figure 3. Scheme of the main autophagy changes related to glaucoma. IOP: intraocular pressure; TM cells: trabecular meshwork cells; RGCs: retinal ganglion cells. (Created in part with
Figure 1. Scheme of the main autophagy changes related to glaucoma. IOP: intraocular pressure; TM cells: trabecular meshwork cells; RGCs: retinal ganglion cells. (Created in part with
, accessed date 20 February 2021).
3.2. Cataract
2.2. Cataract
Autophagic vesicles containing mitochondria or fragments of mitochondria have been observed in human lens fibers. Costello et al. [76] performed a study to confirm the presence of autophagic activity in the lens and demonstrated its presence. Therefore, autophagy helps to maintain the integrity of the cells by contributing to the maintenance of the physical properties of the lens [76] (
Autophagic vesicles containing mitochondria or fragments of mitochondria have been observed in human lens fibers. Costello et al. [25] performed a study to confirm the presence of autophagic activity in the lens and demonstrated its presence. Therefore, autophagy helps to maintain the integrity of the cells by contributing to the maintenance of the physical properties of the lens [25] (
Figure 4). Disturbances in the elimination of organelles in the cytoplasm of lens fibers increase ROS, altering the homeostasis of the lens, decreasing its transparency and leading to the development of cataracts [77] (
). Disturbances in the elimination of organelles in the cytoplasm of lens fibers increase ROS, altering the homeostasis of the lens, decreasing its transparency and leading to the development of cataracts [26] (
Figure 4).
2).
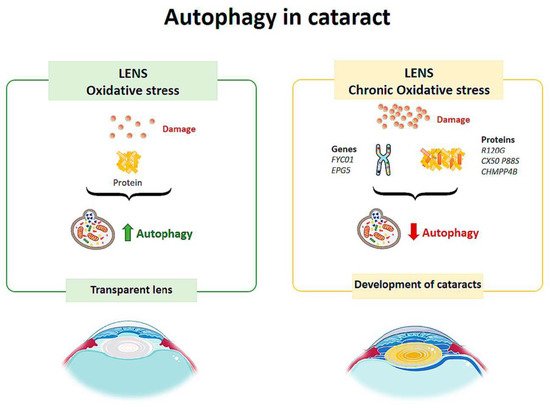
Figure 42.
Scheme of the main autophagy changes related to cataract. (Created in part with
smart.servier.com
, accessed date 20 February 2021).
TBC1D20 (TBC1 domain family member 20) is a key regulator of autophagosome maturation. There is evidence that regulation by TBC1D20 maintains lens transparency. A study conducted on mice tested what would happen if TBC1D20-mediated autophagic flux was altered [78] and found that the animals presented cataracts in the lens nucleus, but the cortex remained transparent. Over time, the pathology progressed, and cataracts completely occupied the lens. In addition, it was observed that the lens fibers were highly degenerated and disorganized, and there was an accumulation of ubiquitin aggregates. Therefore, alteration of the autophagic flux mediated by TBC1D20 resulted in the accumulation of autophagic material in the lens fibers and the formation of cataracts [78]. In another study, Atg5 (autophagy-related protein) knock-out mice showed opacity in the cortical region of the lens after 6–9 months, and by 21 months they had cataracts [79]. The lenses of these mice showed age-dependent cellular disorganization and accumulation of abnormal materials. Accumulation of insoluble p62, ubiquitin aggregates and oxidized proteins was also observed in the lens fibers [79]. These results suggest that autophagy is critical for maintaining intracellular homeostasis in the lens.
TBC1D20 (TBC1 domain family member 20) is a key regulator of autophagosome maturation. There is evidence that regulation by TBC1D20 maintains lens transparency. A study conducted on mice tested what would happen if TBC1D20-mediated autophagic flux was altered [27] and found that the animals presented cataracts in the lens nucleus, but the cortex remained transparent. Over time, the pathology progressed, and cataracts completely occupied the lens. In addition, it was observed that the lens fibers were highly degenerated and disorganized, and there was an accumulation of ubiquitin aggregates. Therefore, alteration of the autophagic flux mediated by TBC1D20 resulted in the accumulation of autophagic material in the lens fibers and the formation of cataracts [27]. In another study, Atg5 (autophagy-related protein) knock-out mice showed opacity in the cortical region of the lens after 6–9 months, and by 21 months they had cataracts [28]. The lenses of these mice showed age-dependent cellular disorganization and accumulation of abnormal materials. Accumulation of insoluble p62, ubiquitin aggregates and oxidized proteins was also observed in the lens fibers [28]. These results suggest that autophagy is critical for maintaining intracellular homeostasis in the lens.
3.3. Diabetic Retinopathy
2.3. Diabetic Retinopathy
In diabetic patients, at the cellular level, hyperglycemia causes the activation of metabolic pathways, resulting in an increase in reactive oxygen species, the appearance of advanced glycation end products, and the release of proinflammatory cytokines, all of which lead to cell death mediated by apoptosis, necroptosis, and autophagy [85]. Therefore, the retinal damage suffered by diabetic patients is related to autophagic events [66].
In diabetic patients, at the cellular level, hyperglycemia causes the activation of metabolic pathways, resulting in an increase in reactive oxygen species, the appearance of advanced glycation end products, and the release of proinflammatory cytokines, all of which lead to cell death mediated by apoptosis, necroptosis, and autophagy [29]. Therefore, the retinal damage suffered by diabetic patients is related to autophagic events [30].
Autophagy is responsible for removing damaged proteins, providing a defense mechanism against RPE damage, but under conditions of excess metabolic stress autophagic activity is impaired [85] (
Autophagy is responsible for removing damaged proteins, providing a defense mechanism against RPE damage, but under conditions of excess metabolic stress autophagic activity is impaired [29] (
Figure 5). High glucose levels also affect Müller cells, which are the major source of VEGF (Vascular Endothelial Growth Factor) in DR. In a study on the effect of hyperglycemia on the retina, rat retinal Müller cells were treated with normal or high glucose levels with/without inhibitors and activators for p62 for 24 hours [86]. Cells in contact with high glucose levels responded with increased autophagic activity and endoplasmic reticulum stress associated with increased apoptosis. By inhibiting autophagy in cells treated with high glucose levels, there was an increased number of Müller cells in apoptosis [86]. In a diabetic mouse model, the physiological autophagic flux was observed to be altered, with increased LC3 at the level of the outer plexiform layer and up-regulation of the autophagic proteins Beclin-1 and Atg5 [87].
3). High glucose levels also affect Müller cells, which are the major source of VEGF (Vascular Endothelial Growth Factor) in DR. In a study on the effect of hyperglycemia on the retina, rat retinal Müller cells were treated with normal or high glucose levels with/without inhibitors and activators for p62 for 24 hours [31]. Cells in contact with high glucose levels responded with increased autophagic activity and endoplasmic reticulum stress associated with increased apoptosis. By inhibiting autophagy in cells treated with high glucose levels, there was an increased number of Müller cells in apoptosis [31]. In a diabetic mouse model, the physiological autophagic flux was observed to be altered, with increased LC3 at the level of the outer plexiform layer and up-regulation of the autophagic proteins Beclin-1 and Atg5 [32].
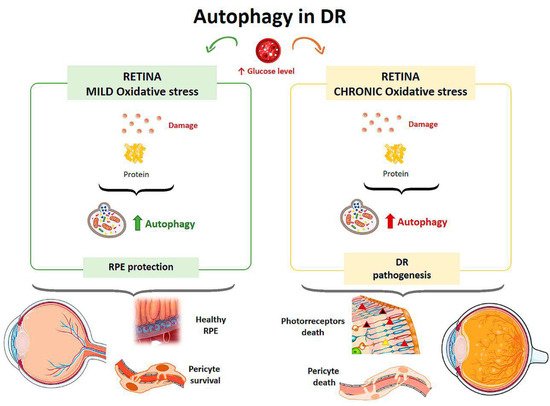
Figure 53.
Scheme of the main autophagy changes related to diabetic retinopathy (DR). RPE: retinal pigment epithelium. (Created in part with
smart.servier.com
, accessed date 20 February 2021).
The effect of hyperglycemia on autophagic activity has been demonstrated in cultured human RPE cells [88]. Cultures were incubated in media with a normal glucose concentration (5 mM) and media with a high glucose concentration (30 mM). Cells treated with high glucose levels showed an increased amount of double membrane vacuoles, typical of autophagosomes, at 48 hours after incubation compared with the culture with a normal glucose concentration. In addition, after exposure to hyperglycemic conditions, cells showed higher levels of LC3 in the cytoplasm, indicating an elevated autophagic response. Autophagy induced by high glucose levels was found to be mTOR-dependent but mainly regulated by ROS-mediated endoplasmic reticulum stress signals. Therefore, under a scenario of oxidative stress induced by high glucose levels, autophagy is required to eliminate damaged proteins and provide a mechanism to prevent RPE induced damage [88] (
The effect of hyperglycemia on autophagic activity has been demonstrated in cultured human RPE cells [33]. Cultures were incubated in media with a normal glucose concentration (5 mM) and media with a high glucose concentration (30 mM). Cells treated with high glucose levels showed an increased amount of double membrane vacuoles, typical of autophagosomes, at 48 hours after incubation compared with the culture with a normal glucose concentration. In addition, after exposure to hyperglycemic conditions, cells showed higher levels of LC3 in the cytoplasm, indicating an elevated autophagic response. Autophagy induced by high glucose levels was found to be mTOR-dependent but mainly regulated by ROS-mediated endoplasmic reticulum stress signals. Therefore, under a scenario of oxidative stress induced by high glucose levels, autophagy is required to eliminate damaged proteins and provide a mechanism to prevent RPE induced damage [33] (
Figure 5). Compared to a control culture, cells that were treated with a high concentration of glucose and were deficient in autophagic processes exhibited a strong reduction in cell viability, suggesting that autophagy plays a protective role for RPE cells in the presence of a DR-common environment [88].
3). Compared to a control culture, cells that were treated with a high concentration of glucose and were deficient in autophagic processes exhibited a strong reduction in cell viability, suggesting that autophagy plays a protective role for RPE cells in the presence of a DR-common environment [33].
3.4. Age-Related Macular Degeneration
Mitochondria from human RPE cells exposed to blue light release ROS, resulting in increased damage to the mitochondrial DNA of these cells [96]. Endogenous ROS generation induces autophagy, and it has been observed that, under conditions of oxidative stress, RPE cells in AMD patients generate elevated levels of ROS compared with healthy RPE cells [94]. When oxidative stress occurs continuously over time, mitophagy is activated as a mechanism to protect the cell against this stress. RPE cell dysfunction in AMD donor samples has been linked to mitochondrial DNA injury, and as the disease severity increases, there is greater damage and reduced ability to repair [97]. Therefore, selective removal of damaged mitochondria by autophagy (mitophagy) could be essential for cell survival (
Mitochondria from human RPE cells exposed to blue light release ROS, resulting in increased damage to the mitochondrial DNA of these cells [34]. Endogenous ROS generation induces autophagy, and it has been observed that, under conditions of oxidative stress, RPE cells in AMD patients generate elevated levels of ROS compared with healthy RPE cells [35]. When oxidative stress occurs continuously over time, mitophagy is activated as a mechanism to protect the cell against this stress. RPE cell dysfunction in AMD donor samples has been linked to mitochondrial DNA injury, and as the disease severity increases, there is greater damage and reduced ability to repair [36]. Therefore, selective removal of damaged mitochondria by autophagy (mitophagy) could be essential for cell survival (
Figure 6).
4).
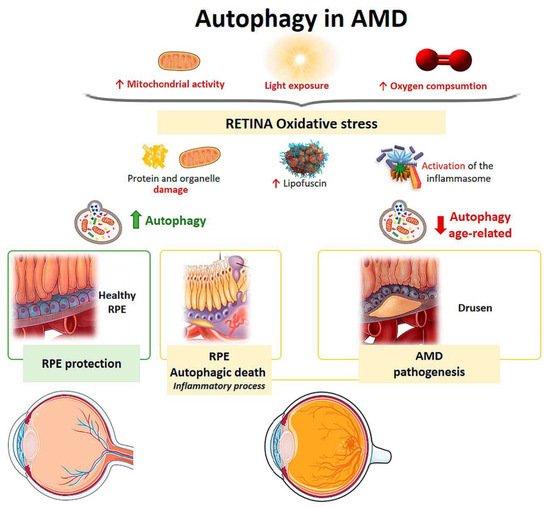
Figure 64.
Scheme of the main autophagy changes related to age related macular degeneration (AMD). RPE: retinal pigment epithelium. (Created in part with
smart.servier.com
, accessed date 20 February 2021).
The retina is under intense oxidative stress due to high oxygen consumption, light exposure and high mitochondrial activity leading to the release of ROS in the RPE. To defend themselves, these cells increase the production of anti-oxidants, which decrease with aging, causing severe oxidative damage and progression of AMD. Oxidative stress can be induced by H
2
O
2
. In vitro models have shown that autophagy-associated cell death occurs in ARPE-19 cells and primary human RPE cells by serum deprivation, and H
2
O
2 (2 h, 1 mM) [98,99]. The phenomenon means, firstly, an induction of a high level of autophagy, which is the cause of cell death, thus autophagic death occurs. Furthermore, these RPE cells undergoing autophagy-associated cell death participate in elimination mechanisms guided by professional and nonprofessional phagocytes and are accompanied by the induction of inflammation. Therefore, this process may be involved in the pathogenesis of AMD [98,99] (
(2 h, 1 mM) [37][38]. The phenomenon means, firstly, an induction of a high level of autophagy, which is the cause of cell death, thus autophagic death occurs. Furthermore, these RPE cells undergoing autophagy-associated cell death participate in elimination mechanisms guided by professional and nonprofessional phagocytes and are accompanied by the induction of inflammation. Therefore, this process may be involved in the pathogenesis of AMD [37][38] (
Figure 6).
4).
Retinal neovascularization may be related to oxidative stress, pyroptosis (a highly inflammatory form of programmed cell death), and impaired autophagy [100]. Another pathological feature of AMD is the accumulation of lipofuscin in the lysosomes of RPE cells [101]. Lipofuscin is a heterogeneous mixture of lipoproteins that sensitize RPE cells to light exposure and oxidative stress, leading to apoptosis [32,86,102]. A2E (N-retinylidene-N-retinyl-ethanolamine) is a lipofuscin fluorophore that is toxically deposited in the RPE cells of AMD patients. Zhang et al. found that A2E triggers autophagy in RPE cells during the early stages of AMD. Inhibition of autophagy in RPE cells with A2E results in elevated levels of proinflammatory and proangiogenic factors [103]. Therefore, an increase in autophagic activity protects against the harmful effects of A2E.
Retinal neovascularization may be related to oxidative stress, pyroptosis (a highly inflammatory form of programmed cell death), and impaired autophagy [39]. Another pathological feature of AMD is the accumulation of lipofuscin in the lysosomes of RPE cells [40]. Lipofuscin is a heterogeneous mixture of lipoproteins that sensitize RPE cells to light exposure and oxidative stress, leading to apoptosis [41][31][42]. A2E (N-retinylidene-N-retinyl-ethanolamine) is a lipofuscin fluorophore that is toxically deposited in the RPE cells of AMD patients. Zhang et al. found that A2E triggers autophagy in RPE cells during the early stages of AMD. Inhibition of autophagy in RPE cells with A2E results in elevated levels of proinflammatory and proangiogenic factors [43]. Therefore, an increase in autophagic activity protects against the harmful effects of A2E.
Aging is the main risk factor for developing AMD. In a study on mouse retinas, autophagy levels were compared at different ages [115]. After lysosomal blockade, aged retinas exhibited lipofuscin-laden lysosomes and ubiquitin aggregates characteristic of autophagic impairment. It was shown that chaperone-mediated up-regulation of autophagy would lead to increased lysosome-dependent proteolysis in aged retinas and that there was uncoordination between autophagic pathways in cone-type photoreceptors. The study proposed that this lack of coordination between the different autophagic pathways could be responsible for the specific pattern of visual loss that occurs in aging (
Aging is the main risk factor for developing AMD. In a study on mouse retinas, autophagy levels were compared at different ages [44]. After lysosomal blockade, aged retinas exhibited lipofuscin-laden lysosomes and ubiquitin aggregates characteristic of autophagic impairment. It was shown that chaperone-mediated up-regulation of autophagy would lead to increased lysosome-dependent proteolysis in aged retinas and that there was uncoordination between autophagic pathways in cone-type photoreceptors. The study proposed that this lack of coordination between the different autophagic pathways could be responsible for the specific pattern of visual loss that occurs in aging (
Figure 6).
4).
2.5. Autophagy as a Therapy in Ocular Pathologies
In recent years, the efficacy of certain drugs that utilize autophagy processes in the treatment of different ocular pathological processes has been proven. These drugs include Rapamycin (RAP), AMPK activator, proteasome inhibitor (MG-132), chloroquine, and hydroxychloroquine, among others [116][45].
In most ocular diseases, proteolytic and autophagic capacity is attenuated. Therefore, some pharmacological activators of autophagy are being studied as potential therapies for ocular pathologies.
A drug that acts as a regulator of the autophagy process is Rapamycin, and it is thought to have a potential therapeutic effect in AMD. It acts through the inhibition of mTOR, which leads to an increase in the autophagy process, protecting RPE cells from A2E toxicity and increasing cell viability [103][43]. In addition to increasing autophagic flux by inhibiting the mTOR pathway, rapamycin treatment, in conjunction with caloric restriction, has been shown to exert a neuroprotective effect on RGCs following transient IOP increase in experimental models of ischemia-reperfusion [73][22]. This neuroprotective effect of rapamycin has also been observed in animal models of optic nerve axotomy [65][46] and in models of chronic glaucoma [117][47].
Resveratrol (natural phenol) and MG-132 (proteasome inhibitor) provide survival stimuli to RPE cells through autophagic induction, so they could be used to prolong the lifetime of RPE cells in AMD [116][45]. AMPK (AMP-activated protein kinase) is an inhibitor of mTOR, so AMPK-mTOR could be a good target for treating AMD [118][48]. Metformin regulates AMP kinase activity, which stimulates the initiation of autophagy [119][49]. Other substances, such as lithium and valproic acid, stimulate the induction of autophagy [120,121][50][51].
The autophagy process could also be stimulated by using microRNAs. These are endogenous small RNAs that regulate gene expression at the post-transcriptional level. Cai et al. showed that the overexpression of micro-RNA-29 (miR-29) in RPE cells could rescue degenerating cells by enhancing autophagy through inhibiting the activity of mTORC1 [122][52].
Hormones such as 17β-Estradiol and melatonin could promote the autophagy process. Wei et al. 2018 found that 17β-estradiol (βE-2) enhanced autophagy and protected RPE cells from oxidative stress induced by a blue light emitting diode (LED) [123][53]. Melatonin, a neurohormone derived from tryptophan, which has crucial effects on several systems (circadian rhythm, aging, immune system, and cardiovascular system), as well as being a potent antioxidant, has been shown to positively regulate the autophagy process [95][54]. It has been shown that in RPE cells exposed to H2O2 (potent inducer of oxidative damage) and treated with melatonin, there was an increase in autophagy mediated by the up-regulation of LC3-II and Beclin-1 and down-regulation of P62 [124][55].
Chloroquine and hydroxychloroquine suppress autophagy by blocking autophagosome fusion and degradation of autophagic contents. Although some studies have shown that these drugs can be effective and even safe, other studies have shown that these agents can bind to the melanin of the RPE increasing the toxic effects of these drugs. Therefore, treatment with chloroquine and hydroxychloroquine may result in retinal toxicity [116][45].
Sodium trans-hinone IIA sulfonate (STS) has been shown to reduce the expression of the autophagic proteins Atg3, Atg7, and Atg9 by inhibiting vacuole elongation and autophagosome formation. In RPE cells, under oxidative stress conditions, STS activates the mTOR/AKT/PI3K pathway, decreasing autophagy-related cell death [127][56].
Although the effectiveness of drugs that act on autophagy processes is promising, these drugs nevertheless have some associated problems. They may have undesirable side effects [128[57][58],129], as they may have more than one route of action, and they may also have low specificity [128][57]. Therefore, there is still much to be investigated to eliminate these problems and to obtain safe and effective drugs that can treat ocular pathologies by regulating autophagy processes.
References
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937.
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995.
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41.
- de Duve, C.; Wattiaux, R. Functions of Lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492.
- Cadwell, K. Crosstalk between autophagy and inflammatory signalling pathways: Balancing defence and homeostasis. Nat. Rev. Immunol. 2016, 16, 661–675.
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–312.
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174.
- Viiri, J.; Amadio, M.; Marchesi, N.; Hyttinen, J.M.T.; Kivinen, N.; Sironen, R.; Rilla, K.; Akhtar, S.; Provenzani, A.; D’Agostino, V.G.; et al. Autophagy Activation Clears ELAVL1/HuR-Mediated Accumulation of SQSTM1/p62 during Proteasomal Inhibition in Human Retinal Pigment Epithelial Cells. PLoS ONE 2013, 8, e69563.
- Wang, A.L.; Lukas, T.J.; Yuan, M.; Du, N.; Tso, M.O.; Neufeld, A.H. Autophagy, exosomes and drusen formation in age-related macular degeneration. Autophagy 2009, 5, 563–564.
- Kim, S.H.; Munemasa, Y.; Kwong, J.M.K.; Ahn, J.H.; Mareninov, S.; Gordon, L.K.; Caprioli, J.; Piri, N. Activation of autophagy in retinal ganglion cells. J. Neurosci. Res. 2008, 86, 2943–2951.
- Fu, C.T.; Sretavan, D. Involvement of EphB/ephrin-B signaling in axonal survival in mouse experimental glaucoma. Investig. Ophthalmol. Vis. Sci. 2012, 53, 76–84.
- Chen, Y.; Sawada, O.; Kohno, H.; Le, Y.Z.; Subauste, C.; Maeda, T.; Maeda, A. Autophagy protects the retina from light-induced degeneration. J. Biol. Chem. 2013, 288, 7506–7518.
- McMonnies, C.W. Hyperbaric oxygen therapy and the possibility of ocular complications or contraindications. Clin. Exp. Optom. 2015, 98, 122–125.
- Pinazo-Duran, M.D.; Shoaie-Nia, K.; Zanon-Moreno, V.; Sanz-Gonzalez, S.M.; del Castillo, J.B.; Garcia-Medina, J.J. Strategies to Reduce Oxidative Stress in Glaucoma Patients. Curr. Neuropharmacol. 2018, 16, 903–918.
- Porter, K.; Nallathambi, J.; Lin, Y.; Liton, P.B. Lysosomal basification and decreased autophagic flux in oxidatively stressed trabecular meshwork cells. Autophagy 2013, 9, 581–594.
- Porter, K.M.; Jeyabalan, N.; Liton, P.B. MTOR-independent induction of autophagy in trabecular meshwork cells subjected to biaxial stretch. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 1054–1062.
- Pulliero, A.; Seydel, A.; Camoirano, A.; Saccà, S.C.; Sandri, M.; Izzotti, A. Oxidative Damage and Autophagy in the Human Trabecular Meshwork as Related with Ageing. PLoS ONE 2014, 9, e98106.
- Porter, K.; Hirt, J.; Stamer, W.D.; Liton, P.B. Autophagic dysregulation in glaucomatous trabecular meshwork cells. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 379–385.
- Wei, T.; Kang, Q.; Ma, B.; Gao, S.; Li, X.; Liu, Y. Activation of autophagy and paraptosis in retinal ganglion cells after retinal ischemia and reperfusion injury in rats. Exp. Ther. Med. 2015, 9, 476–482.
- Piras, A.; Gianetto, D.; Conte, D.; Bosone, A.; Vercelli, A. Activation of Autophagy in a Rat Model of Retinal Ischemia following High Intraocular Pressure. PLoS ONE 2011, 6, e22514.
- Russo, R.; Berliocchi, L.; Adornetto, A.; Varano, G.P.; Cavaliere, F.; Nucci, C.; Rotiroti, D.; Morrone, L.A.; Bagetta, G.; Corasaniti, M.T. Calpain-mediated cleavage of Beclin-1 and autophagy deregulation following retinal ischemic injury in vivo. Cell Death Dis. 2011, 2, e144.
- Russo, R.; Varano, G.P.; Adornetto, A.; Nazio, F.; Tettamanti, G.; Girardello, R.; Cianfanelli, V.; Cavaliere, F.; Morrone, L.A.; Corasaniti, M.T.; et al. Rapamycin and fasting sustain autophagy response activated by ischemia/reperfusion injury and promote retinal ganglion cell survival. Cell Death Dis. 2018, 9, 1–18.
- Hirt, J.; Porter, K.; Dixon, A.; McKinnon, S.; Liton, P.B. Contribution of autophagy to ocular hypertension and neurodegeneration in the DBA/2J spontaneous glaucoma mouse model. Cell Death Discov. 2018, 4, 75.
- Russo, R.; Nucci, C.; Corasaniti, M.T.; Bagetta, G.; Morrone, L.A. Autophagy dysregulation and the fate of retinal ganglion cells in glaucomatous optic neuropathy. Prog. Brain Res. 2015, 220, 87–105.
- Costello, M.J.; Brennan, L.A.; Basu, S.; Chauss, D.; Mohamed, A.; Gilliland, K.O.; Johnsen, S.; Menko, A.S.; Kantorow, M. Autophagy and mitophagy participate in ocular lens organelle degradation. Exp. Eye Res. 2013, 116, 141–150.
- Zhou, J.; Yao, K.; Zhang, Y.; Chen, G.; Lai, K.; Yin, H.; Yu, Y. Thioredoxin Binding Protein-2 Regulates Autophagy of Human Lens Epithelial Cells under Oxidative Stress via Inhibition of Akt Phosphorylation. Oxid. Med. Cell. Longev. 2016, 2016, 1–17.
- Sidjanin, D.J.; Park, A.K.; Ronchetti, A.; Martins, J.; Jackson, W.T. TBC1D20 mediates autophagy as a key regulator of autophagosome maturation. Autophagy 2016, 12, 1759–1775.
- Morishita, H.; Eguchi, S.; Kimura, H.; Sasaki, J.; Sakamaki, Y.; Robinson, M.L.; Sasaki, T.; Mizushima, N. Deletion of Autophagy-related 5 (Atg5) and Pik3c3 Genes in the Lens Causes Cataract Independent of Programmed Organelle Degradation. J. Biol. Chem. 2013, 288, 11436–11447.
- Volpe, C.M.O.; Villar-Delfino, P.H.; dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119.
- Lin, W.; Kuang, H. Oxidative stress induces autophagy in response to multiple noxious stimuli in retinal ganglion cells. Autophagy 2014, 10, 1692–1701.
- Fanjul-Moles, M.L.; López-Riquelme, G.O. Relationship between Oxidative Stress, Circadian Rhythms, and AMD. Oxid. Med. Cell. Longev. 2016, 2016, 1–18.
- Piano, I.; Novelli, E.; Della Santina, L.; Strettoi, E.; Cervetto, L.; Gargini, C. Involvement of autophagic pathway in the progression of retinal degeneration in a mouse model of diabetes. Front. Cell. Neurosci. 2016, 10, 1–12.
- Yao, J.; Tao, Z.F.; Li, C.P.; Li, X.M.; Cao, G.F.; Jiang, Q.; Yan, B. Regulation of autophagy by high glucose in human retinal pigment epithelium. Cell. Physiol. Biochem. 2014, 33, 107–116.
- Godley, B.F.; Shamsi, F.A.; Liang, F.-Q.; Jarrett, S.G.; Davies, S.; Boulton, M. Blue Light Induces Mitochondrial DNA Damage and Free Radical Production in Epithelial Cells. J. Biol. Chem. 2005, 280, 21061–21066.
- Golestaneh, N.; Chu, Y.; Xiao, Y.Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2017, 8, e2537.
- Lin, H.; Xu, H.; Liang, F.-Q.; Liang, H.; Gupta, P.; Havey, A.N.; Boulton, M.E.; Godley, B.F. Mitochondrial DNA Damage and Repair in RPE Associated with Aging and Age-Related Macular Degeneration. Investig. Opthalmol. Vis. Sci. 2011, 52, 3521–3529.
- Szatmári-Tóth, M.; Kristóf, E.; Veréb, Z.; Akhtar, S.; Facskó, A.; Fésüs, L.; Kauppinen, A.; Kaarniranta, K.; Petrovski, G. Clearance of autophagy-associated dying retinal pigment epithelial cells—A possible source for inflammation in age-related macular degeneration. Cell Death Dis. 2016, 7.
- Szatmári-Tóth, M.; Ilmarinen, T.; Mikhailova, A.; Skottman, H.; Kauppinen, A.; Kaarniranta, K.; Kristóf, E.; Lytvynchuk, L.; Veréb, Z.; Fésüs, L.; et al. Human embryonic stem cell-derived retinal pigment epithelium-role in dead cell clearance and inflammation. Int. J. Mol. Sci. 2019, 20, 926.
- Wang, S.; Ji, L.; Li, L.; Li, J. Oxidative stress, autophagy and pyroptosis in the neovascularization of oxygen‑induced retinopathy in mice. Mol. Med. Rep. 2018, 19, 927–934.
- Holz, F.; Schütt, F.; Kopitz, J.; Eldred, G.; Kruse, F.; Völcker, H.; Cantz, M. Inhibition of Lysosomal Degradative Functions in RPE Cells by a Retinoid Component of Lipofuscin. Investig. Ophthalmol. Vis. Sci. 1999, 40, 737–743.
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W.; Ding, J.; et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005.
- De Jong, P.T.V.M. Drusen, AMD, and history. Graefes Arch. Clin. Exp. Ophthalmol. 2015, 253, 2061–2062.
- Zhang, J.; Bai, Y.; Huang, L.; Qi, Y.; Zhang, Q.; Li, S.; Wu, Y.; Li, X. Protective effect of autophagy on human retinal pigment epithelial cells against lipofuscin fluorophore A2E: Implications for age-related macular degeneration. Cell Death Dis. 2015, 6, e1972.
- Rodríguez-Muela, N.; Koga, H.; García-Ledo, L.; de la Villa, P.; de la Rosa, E.J.; Cuervo, A.M.; Boya, P. Balance between autophagic pathways preserves retinal homeostasis. Aging Cell 2013, 12, 478–488.
- Li, Y.; Jiang, Q.; Cao, G.; Yao, J.; Yan, B. Repertoires of Autophagy in the Pathogenesis of Ocular Diseases. Cell. Physiol. Biochem. 2015, 35, 1663–1676.
- Rodríguez-Muela, N.; Germain, F.; Mariño, G.; Fitze, P.S.; Boya, P. Autophagy promotes survival of retinal ganglion cells after optic nerve axotomy in mice. Cell Death Differ. 2012, 19, 162–169.
- Su, W.; Li, Z.; Jia, Y.; Zhuo, Y. Rapamycin Is Neuroprotective in a Rat Chronic Hypertensive Glaucoma Model. PLoS ONE 2014, 9, e99719.
- Hyttinen, J.M.T.; Petrovski, G.; Salminen, A.; Kaarniranta, K. 5′-Adenosine Monophosphate-Activated Protein Kinase–Mammalian Target of Rapamycin Axis As Therapeutic Target for Age-Related Macular Degeneration. Rejuvenation Res. 2011, 14, 651–660.
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174.
- Motoi, Y.; Shimada, K.; Ishiguro, K.; Hattori, N. Lithium and Autophagy. ACS Chem. Neurosci. 2014, 5, 434–442.
- Fu, J.; Shao, C.-J.; Chen, F.-R.; Ng, H.-K.; Chen, Z.-P. Autophagy induced by valproic acid is associated with oxidative stress in glioma cell lines. Neuro. Oncol. 2010, 12, 328–340.
- Cai, J.; Zhang, H.; Zhang, Y.; Zhou, Z.; Wu, S. MicroRNA-29 enhances autophagy and cleanses exogenous mutant αB-crystallin in retinal pigment epithelial cells. Exp. Cell Res. 2019, 374, 231–248.
- Wei, Q.; Liang, X.; Peng, Y.; Yu, D.; Zhang, R.; Jin, H.; Fan, J.; Cai, W.; Ren, C.; Yu, J. 17β-estradiol ameliorates oxidative stress and blue light-emitting diode-induced retinal degeneration by decreasing apoptosis and enhancing autophagy. Drug Des. Devel. Ther. 2018, 12, 2715–2730.
- Zhang, Z.Y.; Bao, X.L.; Cong, Y.Y.; Fan, B.; Li, G.Y.; Žerovnik, E. Autophagy in Age-Related Macular Degeneration: A Regulatory Mechanism of Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 2896036.
- Chang, C.C.; Huang, T.Y.; Chen, H.Y.; Huang, T.C.; Lin, L.C.; Chang, Y.J.; Hsia, S.M. Protective Effect of Melatonin against Oxidative Stress-Induced Apoptosis and Enhanced Autophagy in Human Retinal Pigment Epithelium Cells. Oxid. Med. Cell. Longev. 2018, 2018, 9015765.
- Han, D.; Wu, X.; Liu, L.; Shu, W.; Huang, Z. Sodium tanshinone IIA sulfonate protects ARPE-19 cells against oxidative stress by inhibiting autophagy and apoptosis. Sci. Rep. 2018, 8, 15137.
- Levine, B.; Packer, M.; Codogno, P. Development of autophagy inducers in clinical medicine. J. Clin. Investig. 2015, 125, 14–24.
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293.