In this entry, new pyrimidine derivatives were designed, synthesized and analyzed in terms of their anticancer properties. The tested compounds were evaluated in vitro for their antitumor activity. The cytotoxic effect on normal human dermal fibroblasts (NHDF) was also determined. According to the results, all the tested compounds exhibited inhibitory activity on the proliferation of all lines of cancer cells (colon adenocarcinoma (LoVo), resistant colon adenocarcinoma (LoVo/DX), breast cancer (MCF-7), lung cancer (A549), cervical cancer (HeLa), human leukemic lymphoblasts (CCRF-CEM) and human monocytic (THP-1)).
- pyrimidine
- anticancer
- 3
- 4-dihydronaphthalen
- 6-hydrazinopyrimidine
- lipophilicity
- QSAR study
- topoisomerase II
- DNA intercalating
1. Introduction
As reported by the WHO, 18 million people are presently living with cancer; over 9 million people died from cancer in 2018. Due to the lack of effective and selectively acting anticancer therapies, these numbers are still increasing. The compounds based on the scaffold of pyrimidine exhibit a broad spectrum of pharmacological activity such as anti-inflammatory [1], antimicrobial [2,3], anti-HIV [2], antidiabetic [4] and anticancer activity [5,6,7]. The most recognized drugs based on analogs of pyrimidines are antibacterial (sulfadiazine, trimethoprim), antiviral (trifluridine, idoxuridine), anti-malarial (sulfadoxine), anti-HIV (Retrovir (zidovudine), stavudine), anti-tuberculosis (viomycin), anticancer (5-fluorouracil) agents. The data also show that the adjunction of a [(dialkylamino)alkyl]amino or (hydroxyalkyl)amino side chain enhances anticancer activity of compounds [8,9,10,11]. There are also studies on novel derivatives with a 3,4-dihydronaphthalen moiety that exhibit anticancer activity [12,13]. Another important chemical group that is present in biologically active agents is hydrazone. It exhibits a broad spectrum of anticancer and antimicrobial activity [14,15,16,17,18,19]. The data obtained from the structure–activity relationships (SAR) analysis indicate that combined structures of pyrimidine, 3,4-dihydronaphthalene and alkylamine might have a synergistic anticancer effect.
As reported by the WHO, 18 million people are presently living with cancer; over 9 million people died from cancer in 2018. Due to the lack of effective and selectively acting anticancer therapies, these numbers are still increasing. The compounds based on the scaffold of pyrimidine exhibit a broad spectrum of pharmacological activity such as anti-inflammatory [1], antimicrobial [2][3], anti-HIV [2], antidiabetic [4] and anticancer activity [5][6][7]. The most recognized drugs based on analogs of pyrimidines are antibacterial (sulfadiazine, trimethoprim), antiviral (trifluridine, idoxuridine), anti-malarial (sulfadoxine), anti-HIV (Retrovir (zidovudine), stavudine), anti-tuberculosis (viomycin), anticancer (5-fluorouracil) agents. The data also show that the adjunction of a [(dialkylamino)alkyl]amino or (hydroxyalkyl)amino side chain enhances anticancer activity of compounds [8][9][10][11]. There are also studies on novel derivatives with a 3,4-dihydronaphthalen moiety that exhibit anticancer activity [12][13]. Another important chemical group that is present in biologically active agents is hydrazone. It exhibits a broad spectrum of anticancer and antimicrobial activity [14][15][16][17][18][19]. The data obtained from the structure–activity relationships (SAR) analysis indicate that combined structures of pyrimidine, 3,4-dihydronaphthalene and alkylamine might have a synergistic anticancer effect.
In the present paper, we analyzed and described the anticancer activity of newly designed compounds, a hybrid of pyrimidine–hydrazone moieties, dihydronaphthalene and alkylamine chain. All pyrimidine derivatives were evaluated in vitro for their antitumor activity against LoVo colon adenocarcinoma, LoVo/DX-resistant colon adenocarcinoma, MCF-7 breast cancer, A549 lung cancer, cervical cancer (HeLa), human leukemic lymphoblasts (CCRF-CEM) and human monocytic (THP-1) cell lines. We also evaluated their chemical and physical properties to determine the ability to be an orally active drug in humans. Basically, several different modes of action of the anticancer drug are recognized.
One of the possibilities is controlling transcription factors and polymerases where drugs interact directly with the protein bound to DNA. Another is the non-covalent binding of small molecules to double DNA structure by intercalation or interaction with the minor groove of nucleic acids. Based on the results of QSAR (Quantitative structure–activity relationship) studies, we propose the mode of action of designed compounds by binding to topoisomerase IIα (Topo IIα) in complex with DNA. Our study predicted their binding manner and the binding affinity towards Topo IIα by using molecular docking. It has been confirmed that Topo IIα is often overexpressed in different types of tumors, especially in the G2/M phase of the cell cycle and can be useful as a diagnostic marker [20,21]. Their inhibition leads to DNA double-strand breaks and apoptosis. To date, the FDA has approved etoposide, doxorubicin and mitoxantrone as Topo II-targeted chemotherapy agents [22].
One of the possibilities is controlling transcription factors and polymerases where drugs interact directly with the protein bound to DNA. Another is the non-covalent binding of small molecules to double DNA structure by intercalation or interaction with the minor groove of nucleic acids. Based on the results of QSAR (Quantitative structure–activity relationship) studies, we propose the mode of action of designed compounds by binding to topoisomerase IIα (Topo IIα) in complex with DNA. Our study predicted their binding manner and the binding affinity towards Topo IIα by using molecular docking. It has been confirmed that Topo IIα is often overexpressed in different types of tumors, especially in the G2/M phase of the cell cycle and can be useful as a diagnostic marker [20][21]. Their inhibition leads to DNA double-strand breaks and apoptosis. To date, the FDA has approved etoposide, doxorubicin and mitoxantrone as Topo II-targeted chemotherapy agents [22].
2. Chemistry
The starting point of synthesis was the reaction of 4,6-dichloro-pyrimidine (1) with hydrazine hydrate at room temperature. The resulting compound 4-chloro-6-hydrazinopyrimidine (2) was dissolved in ethanol and reacted with 6-methoxy-1-tetralone to obtain 4-chloro-6-[2-(6-methoxy-3,4-dihydronaphthalen-1(2
H)-ylidene)hydrazinyl]pyrimidine (3)
.Compound 3 was refluxed in a nitrogen atmosphere at normal pressure with
N,
N-diethylethylenediamine or
N,
N-dimethylethylenediamine or 3-amino-1-propanol or
N,
N-dimethyl-1,3-propanediamine, respectively. The synthesis procedure is presented in
Scheme 1(see the Methods section for details).
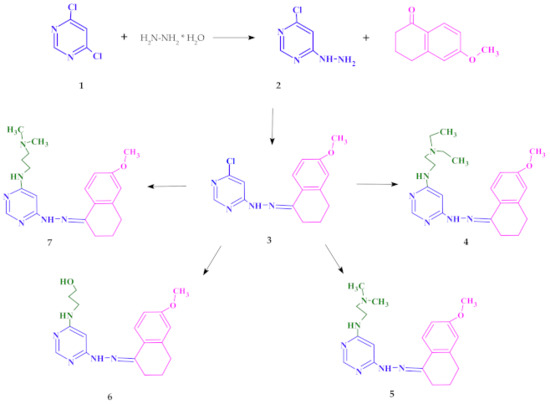
Synthesis of new pyrimidine derivatives.
3. Lipophilicity and QSAR Study
The physicochemical properties like lipophilicity, aqueous solubility, and ADME (absorption, distribution, metabolism and excretion) properties [23] are important for drug candidate substances. While absorption, distribution, metabolism and excretion are related to pharmacokinetics, lipophilicity describes drug interactions with membranes. This is a very important aspect in understanding the mechanisms of transport of substances across the membrane into the cell, especially in the context of multidrug resistance (MDR). The ADME properties could be predicted by using theoretical computational techniques which various companies widely offer. In the presented paper, theoretical physicochemical properties of doxorubicin and all the tested compounds were determined based on the Lipinski and Veber rules on the SwissADME website [24] and were collected in
Table 1. According to the Lipinski’s five rules, all the parameters determined for the tested compounds were in a good agreement with parameters for substances with potentially good pharmacokinetic properties (MW ≤ 500 Da, log P ≤ 5, number of hydrogen bond donors (NHD) ≤ 5 and number of hydrogen bond acceptors (NHA) ≤ 10).
The drug similarity parameters were theoretically obtained from the SwissADME website [24].
NHD—number of hydrogen bond donor;
NHA—number of hydrogen bond acceptors;
NBR—number of rotatable bonds;
TPSA—total polar surface area.
| Compound | Lipinski’s Rules | Veber’s Rules | |||||
|---|---|---|---|---|---|---|---|
| MW ≤ 500 | Log P ≤ 5 | NHD ≤ 5 | NHA ≤ 10 | Violations of Rules | NBR ≤ 10 | TPSA ≤ 140 | |
| Doxorubicin | 543.52 | 0.52 | 6 | 12 | 3 | 5 | 206.07 |
| 4 | 382.50 | 3.14 | 2 | 5 | 0 | 9 | 74.67 |
| 5 | 354.45 | 2.47 | 2 | 5 | 0 | 7 | 74.67 |
| 6 | 341.41 | 2.45 | 3 | 5 | 0 | 7 | 91.66 |
| 7 | 368.48 | 2.77 | 2 | 5 | 0 | 8 | 74.67 |
Unfortunately, the octanol/water partition coefficient, log P
o/wdoes not exhaustively describe lipophilicity but merely quantifies the distribution of the neutral forms of the molecule instead of the distribution coefficient (log D) which includes all electrical species at a given pH. Nevertheless, log P
o/wis an important factor for discovering and developing new effective therapeutics [25]. Among many methods of determining the log P
o/wcoefficient, we chose the shake flask method, which is very simple and allows us to estimate the partition coefficient's relative values in the group of compounds under study. The values obtained for doxorubicin and the synthesized compounds 4, 5 and 7 are summarized in
Table 2. The UV–Vis spectra are shown in
Figure S1(
Supplementary Materials)., could be found in https://www.mdpi.com/1422-0067/22/8/3825).
Experimentally determined log P
values. Shake flask method [26]; P
= [compound]1-oktanol/[compound]HEPES; pH = 7.4; T = 25 °C; concentration determined by UV–Vis absorption. Values of consensus log P
were determined based on the Lipinski and Veber rules on the SwissADME website.
| Compound | Log Po/w | Consensus log Po/w |
|---|---|---|
| Doxorubicin | –0.18 | 0.52 |
| 4 | 1.05 | 3.14 |
| 5 | 1.53 | 2.47 |
| 7 | 1.25 | 2.77 |
Comparing the spectra obtained for standard doxorubicin in a buffer solution at pH 7.4 with the spectra obtained after shaking with n-octanol, the spectra of the aqueous and organic layers were very similar (
Figure S1). This indicates that the form and concentration of doxorubicin were similar in both phases. It is worth mentioning that it is a simple model of an organic/water system that can only account for hydrophobic interactions. This agrees with the fact that about half of the dose is excreted unchanged from the body [27], which might suggest that it does not cross biological membranes. The concentration of the tested compound of the substances in the organic phase significantly exceeded the concentration in the aqueous phase, which is reflected in the log P
o/wvalues. This may explain why the cytotoxic efficacy of the tested compounds compared to doxorubicin was higher. The best extraction into the organic layer was observed in the case of compound 4, which was followed by compounds 7 and 5 (
Figure S1).
The 3D4D/QSAR model with a restricted docking protocol was also used to determine the biological activity of pyrimidine derivatives. We estimated the probability of their inhibitory activity towards topoisomerases I and II. We also analyzed anti-oxidant, DNA anti-metabolic and antimitotic activities. As presented in
Figure 1, compounds 4 and 7 exhibited very high probability of activity towards the Topo II enzyme. Compounds 4, 5 and 7 had antimitotic activity. QSAR analysis also indicated that compound 6 showed high probability of having anti-oxidant and DNA antimetabolic activity.
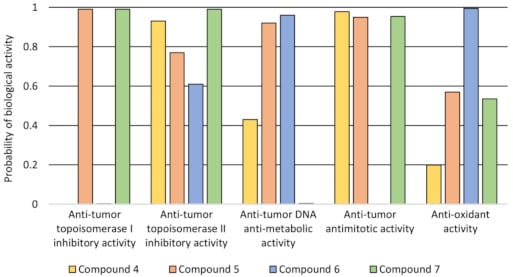
The probability of biological activity predicted based on the 3D/4D QSAR algorithm (0.0–0.2—extremely low, 0.2–0.8—moderate, 0.8–1.0—very high).
4. Biological Evaluation
4.1. Cytotoxicity Assay
The high mortality rate among people with cancer is due to the lack of an effective anticancer drug that selectively targets cancer cells. In this article, we present new 4,6-substituted pyrimidine derivatives with anticancer activity. For a compound to exhibit pharmacological activity, its constituent parts should exhibit pharmacological activity. Properly selected structures contribute to finding a new group exhibiting activity and selectivity and low toxicity to healthy cells. The new structures are combinations of pyrimidine–hydrazone, dihydronaphthalene and alkylamine chain groups. Pyrimidine is a well-known aromatic, heterocyclic organic compound with many different biological functions [3][28][29].
Another functional group present in biologically active agents is hydrazone. It exhibits a broad spectrum of anticancer and antimicrobial activity [14][15][16][17][18][19]. On the other hand, there are reports in the scientific literature about dihydronaphthalene showing biological activity [30][31][32].
The sulforhodamine B (SRB) test was carried out on eight cell lines to characterize cytotoxic properties of the newly synthesized pyrimidine derivatives—one normal and seven cancerous. It was expected to show no cytotoxic effect on cultures of normal cells (normal human dermal fibroblasts (NHDF)) and have cytotoxic effect against tumor cells (cervical cancer (HeLa), breast cancer (MCF7), acute monocytic leukemia (THP-1), adenocarcinoma (LoVo) colorectal carcinoma cell lines—LoVo and LoVo/DX (doxorubicin-resistant), lung cancer (A549) and acute lymphocytic leukemia (CCRF/CEM)). All the tested compounds in the concentration range of 1–20 µM did not reduce the NHDF cells’ viability compared to the control (cultures without tested compounds) and the T0 control (cell culture without the tested compounds’ treatment fixed before adding compounds to the remaining culture plates). Moreover, for compound 7, a statistically significant increase in total cellular protein was observed compared to the control at a concentration of 20 µM. As shown in
B–H, after incubation with all the administered compounds, a reduction in the amount of total protein in all tumor cell cultures was present.
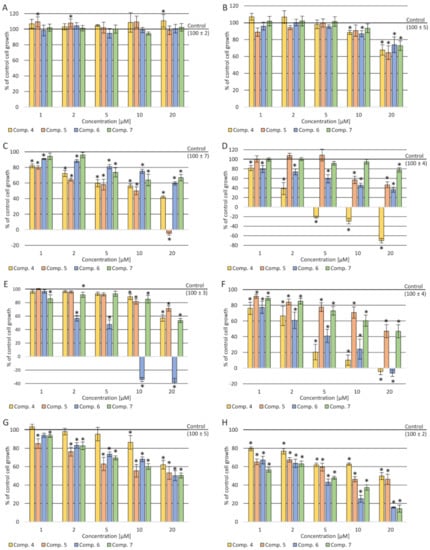
Cytotoxicity effect after incubation of cells with the tested compounds: (
) NHDF cells; (
) HeLa cells; (
) A549 cells; (
) MCF7 cells; (
) CCRF-CEM cells; (
) THP-1 cells; (
) LoVo cells; (
) LoVo/Dx cells. Data presented as a mean and SEM (standard error of the mean); *
< 0.05—significant difference compared to the control.
A decrease in the amount of total protein was observed depending on the concentration used—the higher the concentration, the stronger the cellular protein's inhibition. In the case of THP-1 culture cells, a statistically significant cytotoxic effect of compound 4 was observed in the concentration range of 5–20 µM. In the culture of CCRF-CEM cells, a cytotoxic effect of approx. 8% was demonstrated at a concentration of 20 µM of (5). In a culture of A549 cells, a statistically significant cytotoxic effect was shown in the concentration range of 10–20 µM after incubation with (6). In contrast, in the culture of MCF-7 cells, a slight cytotoxic effect was observed at a concentration of 20 µM after incubation with compounds 4 and 6. In contrast, cell growth inhibition was observed in resistant and sensitive colon cancer cells (stronger in the resistant cell line), but no cytotoxic effect.
4.2. Assessment of the Impact on the Transport Function of P-glycoprotein—Accumulation of Rhodamine 123 (Rod-123) in Cells
Due to the stronger inhibition of the amount of cellular proteins in doxorubicin-resistant colorectal cancer cell cultures than in the counterpart of the cytostatic-sensitive cell line, the activity of P-glycoprotein was tested [11].
Results show a stronger activity inhibiting the compound’s release from LoVo/DX cells compared to doxorubicin for all the tested compounds in the concentration range of 5–20 µM. It was shown that compound 4 strongly influenced the accumulation of rhodamine in cells in the whole range of concentrations tested (
).
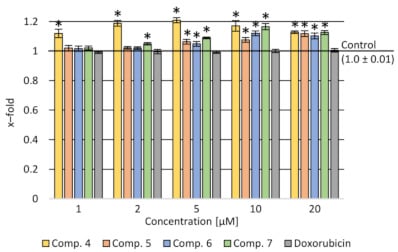
Rhodamine accumulation after incubation with the tested compounds and doxorubicin in various concentration ranges in LoVo/DX cells. Data presented as a mean and SEM (standard error of the mean); *
< 0.05—significant difference compared to the control.
4.3. Verification of Apoptotic and Necrotic Cell Death
The impact of synthesized compounds on neoplastic cell death (A549 lung cancer and CCRF-CEM leukemia) was tested. It was observed that all the tested compounds had a statistically significant effect on cell death as a result of apoptosis (both apoptosis and late apoptosis) (
). Particularly noteworthy is that compound 6 in concentrations of 5 and 10 µM increased the number of cells in the apoptotic phase to over 70%. It is about 20% more than after using the standard drug—doxorubicin. A similar but much smaller effect was seen in the CCRF-CEM cell culture. These cells are characterized by the presence of the mutant p53 protein. The increase of the number of cells in the apoptotic phase was greater at 5 and 10 µM concentrations of (6) than at the corresponding doxorubicin concentrations.
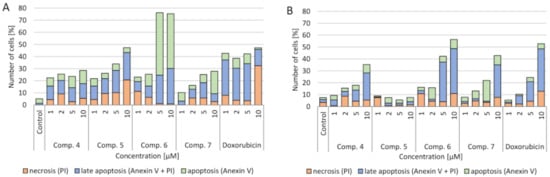
Effect of tested compounds and doxorubicin (Dox) on the level of apoptotic cells (apoptosis, late apoptosis and necrosis after 24 h of incubation): (
) A549 cell line; (
) CCRF-CEM cell line. Apoptosis—annexin V (conjugated with fluorescein) combines with phosphatidylserine, which as a result of damage is located on the outer side of the cell membrane (the color of the cell is green); late apoptosis—when the continuity of the cell membrane has been broken and propidium iodide has entered the cell (the cell has two colors—green and red); necrosis—when the membrane is degraded and the cell stains only red as a result of propidium iodide fusion with the cell nucleus.
4.4. Cell Cycle
The influence of the tested compounds on the cell cycle was also checked. In both A549 and CCRF-CEM cell cultures, a concentration-dependent increase in the number of cells in the G2/M phase after incubation with doxorubicin was observed. On the other hand, after applying the tested compounds, an increase in the number of cells in the G1 phase and a decrease in the proliferation (S) phase in both lung cancer and leukemia cells were observed (
).
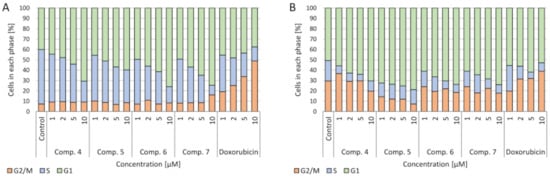
Effect of the tested compounds and doxorubicin (Dox) on the cell cycle after 24 h of incubation: (
) CCRF-CEM cell line; (
) A549 cell line.
4.5. Cell Migration
A serious problem in the treatment of cancer is the risk of metastasis and rapid uncontrolled tumor growth. Therefore, reduction of the crack growth rate in the monolayer of A549 cell culture was investigated. The change increment rate was calculated during the 24-h cultivation with the tested primers (
). The concentration dependence was demonstrated—the higher the tested compounds’ concentration, the slower the neoplastic cells spread. At the same time, at the concentration of 10 µM, a smaller multiplication of tumor cells was observed than with doxorubicin. The use of 1 µM of all the tested compounds and doxorubicin did not reduce tumor progression than the control. Simultaneously, the calculated scratch surface in the monolayer after 24 h of incubation was the highest at concentrations of 10–5 µM. The strongest inhibition of the spread of neoplastic cells was observed after the use of (6).
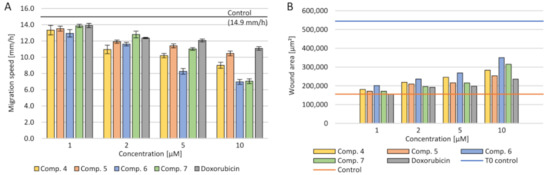
Effect of the tested compounds and doxorubicin (Dox) on the migration of cells in the scratch assay after 24 h of incubation for the A549 cell line: (
) migration speed; (
) wound area after 24 h.
5. Molecular Docking
We performed a molecular docking study to determine the binding mode of synthesized compounds to the Topo II/DNA complex (PDB ID 5GWK). We obtained the free energy of binding (ΔG
binding
) and the inhibition constants (K
i
) (see
Table 3). Additionally, the non-covalent interactions were characterized in detail. The docking protocol was validated by docking the co-crystalized ligand etoposide to the active site of Topo IIα. Our results are consistent with the earlier studies [33,34]. Etoposide binds to the binding site of topoisomerase and DNA with the free energy of binding equal to –44.6 kJ/mol. The aglycone part of the inhibitor localized between nucleic acid bases is involved in drug–DNA interactions (π–π interactions with thymine DT9 and guanine DG13). The podophyllotoxin moiety also contributes to drug–protein interactions and binds to the pocket created by Glu461, Gly462, Asp463, Arg487 and Gly488. The complex is also stabilized by van der Waals interactions and hydrogen bonds created with Gly462, Asp463 and guanine DG13 (see
). Additionally, the non-covalent interactions were characterized in detail. The docking protocol was validated by docking the co-crystalized ligand etoposide to the active site of Topo IIα. Our results are consistent with the earlier studies [33][34]. Etoposide binds to the binding site of topoisomerase and DNA with the free energy of binding equal to –44.6 kJ/mol. The aglycone part of the inhibitor localized between nucleic acid bases is involved in drug–DNA interactions (π–π interactions with thymine DT9 and guanine DG13). The podophyllotoxin moiety also contributes to drug–protein interactions and binds to the pocket created by Glu461, Gly462, Asp463, Arg487 and Gly488. The complex is also stabilized by van der Waals interactions and hydrogen bonds created with Gly462, Asp463 and guanine DG13 (see
and
and
).
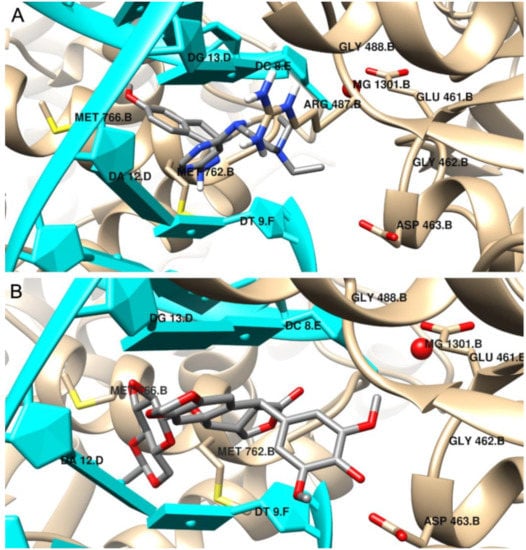
The binding mode of: (
) the most potent inhibitor 4, (
) etoposide in the active site of Topo IIα; 3D representation, DNA structure is marked in cyan, the protein chain is yellow, ligand structures are grey.
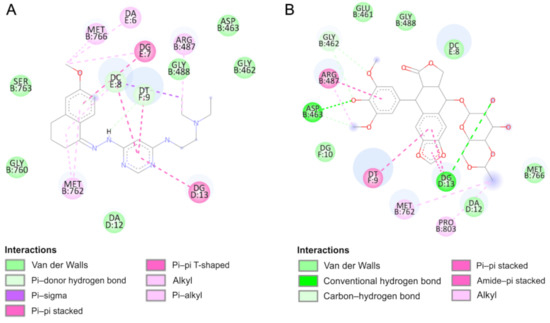
Intermolecular interactions in the active site of Topo IIα (2D representation) of: (
) the most potent inhibitor 4, (
) etoposide.
Free energy of binding, inhibition constant and intermolecular interaction energy components evaluated during molecular docking of one molecule of the inhibitor to the active site of Topo IIα.
| Compound | ΔGbinding (kJ/mol) |
Ki (µM) |
ΔGint (kJ/mol) |
ΔGvdw + ΔGhbond + ΔGdesolv (kJ/mol) |
ΔGel (kJ/mol) |
|---|---|---|---|---|---|
| 4 | −30.8 | 4.12 | −42.0 | −41.6 | −0.4 |
| 5 | −29.0 | 8.56 | −37.7 | −37.5 | −0.2 |
| 6 | −26.3 | 24.36 | −36.3 | −35.5 | −0.8 |
| 7 | −27.0 | 15.77 | −37.0 | −36.8 | −0.2 |
| Etoposide | −44.6 | 0.02 | −50.9 | −50.6 | −0.3 |
According to the results, the most potent inhibitor of Topo IIα is compound 4 (inhibition constant, 4.12 µM). The pyrimidine ring is intercalated in double-stranded DNA and interacts via π–π- interactions with cytosine DC8, thymine DT9 and guanine DG13. The naphthalene moiety is involved in π-stacked interactions with guanine DG7 and π–alkyl interactions with Met762. Additionally, compound 4 forms van der Waals interactions with a number of polar, charged and hydrophobic amino acids of topoisomerase, namely, Gly462, Asp463, Gly488, Gly760 and Ser763.
As can be observed, the geometry of the designed ligands affects their binding properties and inhibitory activity. The data are presented in
and in the
. The
N
,
N
-diethylethylenediamine group with a
N
,
N
-dimethylethylenediamine fragment slightly changes the nature of interactions (compound 5). In this case, the naphthalene ring interacts via π–π stacking interactions with cytosine DC8, thymine DT9 and guanine DG13 and via π–σ interactions with adenine DA12. The van der Waals interactions of Glu461, Glu462, Gly760, Met762 and Tyr805 amino acid residues of Topo II and pyrimidine and
N
,
N
-dimethylethylenediamine fragments are present. The total intermolecular interaction energy for this complex is equal to −37.7 kJ/mol.
The results indicate that compound 6 is the weakest inhibitor of topoisomerase (ΔG
binding
= −26.3 kJ/mol), which is consistent with the quantitative structure–activity relationship study. It is probably related to the replacement of the branched chain of the 3-amino-1-propanol group, which interacts with the protein via hydrogen bonds with Asp463 and van der Waals interactions with Gly462, Ser464 and Gly488 amino acid residues. The naphthalene moiety exhibits a unique binding configuration, namely, interacts through π–σ interactions with guanine DG13, via a π–π stacked configuration with thymine DT9 and via π–alkyl interaction with adenine DA12. Its mechanisms of action can probably be different. QSAR studies showed that it has potential anti-oxidant and DNA antimetabolic activity. On the other hand, an in vitro study showed its strong proapoptotic properties.
Compound 7 can form hydrogen bonds with guanine molecules (DG10 and DG13). Additionally, the naphthalene ring is directly exposed to π–π interactions with thymine DT9 and guanine DG13. One alkyl interaction was observed between
N
,
N
-dimethyl-propanediamine chain and the Lys440 residue. In this case, three carbon–hydrogen bonds and van der Waals interactions were detected with Asp463, Pro485, Glu506 and Gly462 and Leu486, respectively.
References
- Kaur, H.; Machado, M.; de Kock, C.; Smith, P.; Chibale, K.; Prudêncio, M.; Singh, K. Primaquine-pyrimidine hybrids: Synthesis and dual-stage antiplasmodial activity. Eur. J. Med. Chem. 2015, 101, 266–273.
- Desai, N.C.; Kotadiya, G.M.; Trivedi, A.R. Studies on molecular properties prediction, antitubercular and antimicrobial activities of novel quinoline based pyrimidine motifs. Bioorgan. Med. Chem. Lett. 2014, 24, 3126–3130.
- Su, L.; Li, J.; Zhou, Z.; Huang, D.; Zhang, Y.; Pei, H.; Guo, W.; Wu, H.; Wang, X.; Liu, M.; et al. Corrigendum to “Design, synthesis and evaluation of hybrid of tetrahydrocarbazole with 2,4-diaminopyrimidine scaffold as antibacterial agents” [Eur. J. Med. Chem. 162 (162) (2019) 203–211]. Eur. J. Med. Chem. 2019, 168, 385.
- Barakat, A.; Soliman, S.M.; Al-Majid, A.M.; Lotfy, G.; Ghabbour, H.A.; Fun, H.K.; Yousuf, S.; Choudhary, M.I.; Wadood, A. Synthesis and structure investigation of novel pyrimidine-2,4,6-trione derivatives of highly potential biological activity as anti-diabetic agent. J. Mol. Struct. 2015, 1098, 365–376.
- Zimmermann, J. Pyrimidine Derivatives and Processes for the Preparation Thereof. U.S. Patent US5521184A, 28 May 1996.
- Xie, F.; Zhao, H.; Zhao, L.; Lou, L.; Hu, Y. Synthesis and biological evaluation of novel 2,4,5-substituted pyrimidine derivatives for anticancer activity. Bioorgan. Med. Chem. Lett. 2009, 19, 275–278.
- Kaldrikyan, M.A.; Grigoryan, L.A.; Geboyan, V.A.; Arsenyan, F.G.; Stepanyan, G.M.; Garibdzhanyan, B.T. Synthesis and antitumor activity of some disubstituted 5-(3-methyl-4-alkoxybenzyl)pyrimidines. Pharm. Chem. J. 2000, 34, 521–524.
- Tylinska, B.; Jasztold-Howorko, R.; Kowalczewska, K.; Szczaurska-Nowak, K.; Gbarowski, T.; Wietrzyk, J. Design, synthesis and analysis of anticancer activity of new SAR-based S16020 derivatives. Acta Pol. Pharm. Drug Res. 2018.
- Jasztold-Howorko, R.; Tylińska, B.; Biaduń, B.; Gębarowski, T.; Gąsiorowski, K. New pyridocarbazole derivatives. Synthesis and their in vitro anticancer activity. Acta Pol. Pharm. 2013, 70, 823–832.
- Nguyen, C.H.; Bisagni, E.; Lhoste, J.M.; Lavelle, F.; Bissery, M.C. Synthesis and Antitumor Activity of l-[[(Dialkylamino)alkyl]amino]-4-methyl-5H-pyrido[4,3-b]benzo[e]- and -benzo[g])indoles. A New Class of Antineoplastic Agents. J. Med. Chem. 1990, 33, 1519–1528.
- Piasny, J.; Wiatrak, B.; Dobosz, A.; Tylińska, B.; Gębarowski, T. Antitumor Activity of New Olivacine Derivatives. Molecules 2020, 25, 2512.
- Wang, F.; Zhang, R.; Cui, Y.; Sheng, L.; Sun, Y.; Tian, W.; Liu, X.; Liang, S. Design, synthesis and biological evaluation of 3,4-dihydronaphthalen-1(2H)-one derivatives as Bcl-2 inhibitors. Res. Chem. Intermed. 2017, 43, 5933–5942.
- Ananth, A.H.; Manikandan, N.; Rajan, R.K.; Elancheran, R.; Lakshmithendral, K.; Ramanathan, M.; Bhattacharjee, A.; Kabilan, S. Design, Synthesis, and Biological Evaluation of 2-(2-Bromo-3-nitrophenyl)-5-phenyl-1,3,4-oxadiazole Derivatives as Possible Anti-Breast Cancer Agents. Chem. Biodivers. 2020, 17.
- Katariya, K.D.; Shah, S.R.; Reddy, D. Anticancer, antimicrobial activities of quinoline based hydrazone analogues: Synthesis, characterization and molecular docking. Bioorgan. Chem. 2020, 94, 103406.
- Horiuchi, T.; Chiba, J.; Uoto, K.; Soga, T. Discovery of novel thieno[2,3-d]pyrimidin-4-yl hydrazone-based inhibitors of Cyclin D1-CDK4: Synthesis, biological evaluation, and structure-activity relationships. Bioorgan. Med. Chem. Lett. 2009, 19, 305–308.
- Pandey, J.; Pal, R.; Dwivedi, A.; Hajela, K. Synthesis of some new diaryl and triaryl hydrazone derivatives as possible estrogen receptor modulators. Arzneimittelforschung Drug Res. 2002, 52, 39–44.
- Kaplancıklı, Z.A.; Yurttaa, L.; Turan-Zitouni, G.; Özdemir, A.; Göger, G.; Demirci, F.; Mohsen, U.A. Synthesis and Antimicrobial Activity of New Pyrimidine-Hydrazones. Lett. Drug Des. Discov. 2014, 11, 76–81.
- Nasr, T.; Bondock, S.; Rashed, H.M.; Fayad, W.; Youns, M.; Sakr, T.M. Novel hydrazide-hydrazone and amide substituted coumarin derivatives: Synthesis, cytotoxicity screening, microarray, radiolabeling and in vivo pharmacokinetic studies. Eur. J. Med. Chem. 2018, 151, 723–739.
- Dweedar, H.E.; Mahrous, H.; Ibrahim, H.S.; Abdel-Aziz, H.A. Analogue-based design, synthesis and biological evaluation of 3-substituted-(methylenehydrazono)indolin-2-ones as anticancer agents. Eur. J. Med. Chem. 2014, 78, 275–280.
- Park, S.-E.; Chang, I.-H.; Jun, K.-Y.; Lee, E.; Lee, E.-S.; Na, Y.; Kwon, Y. 3-(3-Butylamino-2-hydroxy-propoxy)-1-hydroxy-xanthen-9-one acts as a topoisomerase IIα catalytic inhibitor with low DNA damage. Eur. J. Med. Chem. 2013, 69, 139–145.
- Byl, J.A.W.; Cline, S.D.; Utsugi, T.; Kobunai, T.; Yamada, Y.; Osheroff, N. DNA topoisomerase II as the target for the anticancer drug TOP-53: Mechanistic basis for drug action. Biochemistry 2001, 40, 712–718.
- Chikamori, K.; Grozav, A.; Kozuki, T.; Grabowski, D.; Ganapathi, R.; K Ganapathi, M. DNA Topoisomerase II Enzymes as Molecular Targets for Cancer Chemotherapy. Curr. Cancer Drug Targets 2010, 10, 758–771.
- Zhang, M.Q.; Wilkinson, B. Drug discovery beyond the “rule-of-five”. Curr. Opin. Biotechnol. 2007, 18, 478–488.
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13.
- Arnott, J.A.; Planey, S.L. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discov. 2012, 7, 863–875.
- OECD. Test No. 107: Partition Coefficient (n-octanol/water): Shake Flask Method. In OECD Guidelines for the Testing of Chemicals, Section 1; OECD Publishing: Paris, France, 1995.
- Boucek, R.J.; Olson, R.D.; Brenner, D.E.; Ogunbunmi, E.M.; Inui, M.; Fleischer, S. The major metabolite of doxorubicin is a potent inhibitor of membrane-associated ion pumps. A correlative study of cardiac muscle with isolated membrane fractions. J. Biol. Chem. 1987, 262, 15851–15856.
- Vekariya, M.K.; Vekariya, R.H.; Brahmkshatriya, P.S.; Shah, N.K. Pyrimidine-based pyrazoles as cyclin-dependent kinase 2 inhibitors: Design, synthesis, and biological evaluation. Chem. Biol. Drug Des. 2018, 92, 1683–1691.
- Malik, A.; Rasool, N.; Kanwal, I.; Hashmi, M.A.; Zahoor, A.F.; Ahmad, G.; Altaf, A.A.; Shah, S.A.A.; Sultan, S.; Zakaria, Z.A. Suzuki–Miyaura Reactions of (4-bromophenyl)-4,6-dichloropyrimidine through Commercially Available Palladium Catalyst: Synthesis, Optimization and Their Structural Aspects Identification through Computational Studies. Processes 2020, 8, 1342.
- Kirby, A.J.; Le Lain, R.; Maharlouie, F.; Mason, P.; Nicholls, P.J.; John Smith, H.; Simons, C. Inhibition of Retinoic Acid Metabolising Enzymes by 2-(4-aminophenylmethyl)-6-hydroxy-3,4-dihydronaphthalen-1(2H)-one and Related Compounds. J. Enzyme Inhib. Med. Chem. 2003, 18, 27–33.
- Sun, Y.; Zhou, Y.-Q.; Liu, Y.-K.; Zhang, H.-Q.; Hou, G.-G.; Meng, Q.-G.; Hou, Y. Potential anti-neuroinflammatory NF-кB inhibitors based on 3,4-dihydronaphthalen-1(2H)-one derivatives. J. Enzyme Inhib. Med. Chem. 2020, 35, 1631–1640.
- Rath, S.K.; Singh, S.; Kumar, S.; Wani, N.A.; Rai, R.; Koul, S.; Khan, I.A.; Sangwan, P.L. Synthesis of amides from (E)-3-(1-chloro-3,4-dihydronaphthalen-2-yl)acrylic acid and substituted amino acid esters as NorA efflux pump inhibitors of Staphylococcus aureus. Bioorgan. Med. Chem. 2019, 27, 343–353.
- Wu, C.C.; Li, T.K.; Farh, L.; Lin, L.Y.; Lin, T.S.; Yu, Y.J.; Yen, T.J.; Chiang, C.W.; Chan, N.L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459–462.
- Montecucco, A.; Zanetta, F.; Biamonti, G. Molecular mechanisms of etoposide. EXCLI J. 2015, 14, 95–108.