Cancer is a multifactorial disease with increasing incidence. There are more than 100 different cancer types, defined by location, cell of origin, and genomic alterations that influence oncogenesis and therapeutic response. This heterogeneity between tumors of different patients and also the heterogeneity within the same patient’s tumor pose an enormous challenge to cancer treatment.
- intra-tumor heterogeneity
- multi-omics technology
- cancer models
- patient-derived organoids
- personalized oncology
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
Despite all advancements in research and clinical practice, cancer remains a life-threatening disease with increasing incidence. Based on a prognosis by the WHO in 2018, cancer incidence is expected to double to about 37 million new cancer cases by 2040 [1].
Effective disease management is critical to cancer treatment. Current cancer treatments have made enormous progress from the first cytotoxic agents aiming at replicating cells via targeted therapies selectively aiming at genomic aberrant pathways like cetuximab for the treatment of advanced colorectal cancer to immuno-targeted drugs like ipilimumab for the treatment of malignant melanoma [2][3][2,3].
Despite the progress in medical oncology, most cancers are still treated by surgical resection of the tumors if accessible [4]. Pivotal to subsequent adjuvant or neo-adjuvant chemotherapy is the application of strict guideline protocols (S3 guidelines in Europe). These protocols are based on studies of large patient cohorts with similar cancers. This approach has led to a significant increase in progression-free (PFS) and overall survival (OAS) of patients, yet a majority of patients does not fully benefit from the administered treatment regimen [5]. The reason for therapeutic regimens falling short in those patients is the fact that every cancer comes with an individual, virtually unique genetic landscape [6].
Until it will become possible to faithfully predict the individual outcome of a specific treatment, oncologists and patients alike experience painful uncertainty regarding therapy success at the start and course of treatment. Even worse, both parties know that even when the tumor does not regress, the dire side effects of treatment will still impact the patient’s quality of life.
It is the aim of precision oncology to overcome this dilemma. Understanding the interplay between the unique characteristics of a patient’s tumor and the medical treatment and how it can be tailored to the individual properties of the tumor is a major focus in translational cancer research. Today, cancer precision medicine mostly aims at matching specific tumor mutations with drugs targeting aberrant oncogenic pathways to provide individualized treatment options relying on small organic compounds and/or monoclonal antibodies [7]. While most cancers harbor multiple oncogenic mutations, preclinical and clinical data support the idea that many cancers are sensitive to inhibition of single oncogenes, a concept referred to as ‘oncogene addiction’ [8].
This mutation-driven approach to cancer precision medicine is also applied to pre-therapeutic cohort stratification, which has subsequently led to the concept of conditional approvals, i.e., certain, targeted cancer therapeutics are only approved for patients with a defined set of specific mutations.
The precedent of a targeted cancer therapeutic with conditional approval is the anti-epidermal growth factor receptor (EGFR) antibody cetuximab (Erbitux®). In colorectal cancer (CRC), examination of molecular alterations indicated that mutations in KRAS, which is downstream from EGFR in the RAS-MEK-ERK signaling pathway, interfered with this therapy [9][10][9,10]. Cetuximab is therefore only relevant for RAS wildtype tumors. Following the success of cetuximab, other small molecules, biosimilar and monoclonal antibodies were investigated and successfully applied in the clinic. Today, our arsenal of targeted drugs in oncology comprises 84 approved agents [11].
Yet, cancer is far from being conquered, reflected by the fact that cancer is the second leading cause of death worldwide accounting for an estimated 9.6 million deaths in 2018 [12]. In addition, it becomes more and more evident that the genetic approach to cancer precision medicine alone is not sufficient to predict individual treatment response. This is mainly due to intra-tumor heterogeneity, which is currently not sufficiently taken into consideration.
2. Intra-Tumor Heterogeneity—The Challenge of Treating “Many Cancers in One”
The biology of cancer is complex and not yet fully understood. During malignant transformation tumors may acquire increasingly aggressive features and over time increase their metastatic potential and propensity to gain treatment resistance [13][14]. These hallmark features develop by clonal evolution which is fueled by the complex interplay of cancer cells and their microenvironment [15]. This unique composition of any given tumor is one of the biggest clinical challenges in modern oncology [16][17].
An emerging field is the immunogenic heterogeneity, which is common in liver tumors but not limited to this tumor entity [22][23]. Tumors from different patients show a different degree of immune cell infiltration and immune cell composition [23]. Immunologically “hot” tumors present high levels of T cell infiltration [24]. In consequence, these tumors are more susceptible to immune-checkpoint inhibitor treatment as compared to immunologically “cold” tumors [24]. This immunogenic heterogeneity impacts treatment outcome. There are multiple studies ongoing which aim to define a consensus classification for molecular (immune) subclasses [22][25].
Intra-patient heterogeneity or intra-tumor heterogeneity (ITH) has been proven for solid tumors and usually refers to genetic changes within cell subpopulations that form the tumor mass after multiple cell divisions and proliferation during tumor growth [26][27][28][29]. Thus, solid tumors can be described as heterogeneous neoplasms comprised of different types of cells. A representative solid tumor is composed of malignant cells, communized with mesenchymal cells, endovascular cells, and immune cells creating the tumor microenvironment [30].
ITH is the result of rather complex events and context, related to different causes and different outcome patterns. There are different types of ITH: morpho-histological [31][32], clonal [15] and nonclonal ITH [33][34][35].
Nonclonal ITH derives from microenvironment interaction which makes the clinical approach challenging [31]. The tumor microenvironment is in constant chemical and physical interaction with the actual tumor. These interactions vary between the different areas of the primary tumor. Further, these interactions may not only fuel heterogeneity amongst the tumor cells, but also the stroma can become increasingly heterogeneous [33]. Besides paracrine signaling of cancer and stroma cells, the interaction with the different types of collagen within the stroma may impact therapy success [34][35].
Following the development of new analytic technologies, recent data support the clonal evolution model as the main theoretical basis of tumor heterogeneity [19][39].
G12D
1047R
C242F
R361H
The impact of ITH on drug resistance and targeted therapy strategies has been demonstrated using multiple-site profiling in solid tumors and is regarded as a paradigm shift in cancer care [42]. In contrast, only a limited number of studies addressed ITH in non-solid tumors [43][44][45][46]. Wogsland et al. studied intra- and inter-tumor heterogeneity of follicular lymphoma (FL), a B-cell malignancy, by using mass cytometry to obtain deep profiling of cell subsets [43]. This study allowed the identification of biologically relevant features including tumor heterogeneity and loss of non-malignant B-cell subsets. Additional proteins with a high variability among lymphoma cells have been identified in the same tumor [43].
3. Integration of Intra-Tumor Heterogeneity into Multi-Layered Personalized Cancer Therapy
Regardless of the tumor evolution that is causing ITH, the existence of multiple distinctive cell populations in the same tumor has strong clinical implications [48]. The diagnosis of primary and metastatic tumors is typically based on a single biopsy representing only a snapshot of ongoing tumor evolution and may be compromised by ITH [49]. The studies described above show that a sole biopsy does not accurately capture the tumor’s genetic and phenotypic heterogeneity [28][46]. Therefore, multimodal strategies have to be taken into account to ensure patients receive effective and targeted therapy. In addition, it is necessary to develop new tools to study heterogeneity, and to identify new biomarkers of tumor heterogeneity. Further, besides focusing on clonal heterogeneity, nonclonal phenotypic heterogeneity should be taken into consideration, i.e., the fact that some cells respond to broad, environmental perturbations and drug treatments by conversion to many other cell states, including stem-like, resistant cell phenotypes [50]. Finally, suitable models are needed that also take the effect of the tumor microenvironment into account.
3.1. Current Approaches to Analyze ITH from Tumor Samples
3.1.1. Genomic Approaches to ITH and Precision Oncology
The presence of multiple clones within the same tumor sparked an ongoing discussion about the need of multi-regional sampling to discover ITH in tumors. Surprisingly, Zhang et al. concluded that complete assessment of ITH complexity may not require sampling in multiple regions [57]. They showed for lung adenocarcinoma that a single biopsy may be sufficient to capture the majority of mutations if ultra-deep sequencing is performed [57]. This is in stark contrast to other studies like the one by Gerlinger et al. [58]. Multiregional sequencing on samples obtained from primary renal carcinomas and associated metastatic sites demonstrated substantial ITH, with several mutations in certain cancer genes being restricted to separated tumor domains [58]. These studies suggest that a single biopsy may not be suitable for the identification of all cancer gene mutations of a tumor, thus providing an incomplete view of potential targets for cancer therapy [13][58][59].
RAS
RAS
RAS wild-type patients benefit from upstream RAS inhibition due to the tissue context [63][64][65][66][67][68]. Patients with right hemi colic cancers do not benefit from anti-EGFR therapy irrespective of RAS status [69]. Current guidelines take such spatial heterogeneity into account, and therefore suggest the use of cetuximab or panitumumab only for patients with
3.1.2. Proteomic Approaches
During the last few decades, not only have sequencing technologies advanced significantly, but other technologies such as proteome and metabolome analyses have also become more suitable for a deeper characterization of tumor cells and their functional abnormalities [72][73].
Despite the complexity, proteomic approaches have led to the identification of specific biomarkers in ovarian cancer [65] and identification of molecular subgroups in breast cancer [66]. Proteomic data used in precision oncology has helped to correctly predict drug sensitivity and resistance [67][68]. Cancer proteomics can therefore be seen as complementing the traditional immunohistochemical classification of tumor types, such as the characterization of estrogen receptor expression in breast cancers [77][78].
3.1.3. Metabolomic Approaches
Metabolites are the products of cellular processes, which in turn are driven by proteins, mostly enzymes. Therefore, changes in metabolites mirror changes in the activity of enzymes and proteins and may pose as ideal biomarkers [79]. Due to their accessibility, many metabolomic analyses are performed on plasma or serum samples from patients used in diagnosis without requiring an invasive intervention to obtain tumor tissue [80][81]. Yet, this analysis of individual levels of metabolites makes it difficult to determine universal levels and individual changes for a particular tumor entity. Currently, only few studies have been conducted, and validation is pending [82]. Among the few examples of a clinically relevant metabolomic approach is the use of metabolites for the identification of altered carbohydrates in acute myeloid leukemia [83], as well as unsaturated free fatty acids in colorectal cancer [84].
3.2. Suitable In Vitro Strategies for Modeling Intra-Tumor Heterogeneity
Cancer models are naturally existing or artificially induced systems that share characteristics with human cancers [85]. Experimental systems for the study of human cancer include genetically engineered mouse models (GEMMs) [86][87][88][89], two-dimensional (2D) cell lines [90], patient-derived organoids (PDO) [91][92][93][94][95], and patient-derived xenografts (PDX) [95][96] to study biochemical or genetic pathways and pathology of cancer. These in vivo and in vitro cancer models have been invaluable for our current understanding of cancer development and progression, as well as for therapy development. Further, these models are moving into focus regarding their potential use in cancer precision and/or personalized medicine [97][98].
GEMMs are created by inducing specific mutations in oncogenes and/or tumor suppressor genes and can be used to monitor tumorigenesis in vivo, but are limited by species differences in oncogenic pathogenesis, the shorter lifespan of mice, and often by the artificial simultaneous introduction of several oncogenic driver events [88][89].
Traditional 2D cell lines grow as monolayer, cultured on flat and rigid substrates [91]. They have the advantage that they have once been derived from a cancer patient and are easier to manipulate in the laboratory, but they cannot completely replicate the environment of the patient tumor. Even within the same cancer model, data between laboratories are often irreproducible [99][100]. Nevertheless, cancer 2D cell lines have been used not only for in vitro but also for in vivo experiments, for example to generate xenograft models by subcutaneous injection of cancer cell lines into immunodeficient mice [101].
Patient-derived xenografts (PDX) are generated by implantation of cancerous tissue from a patient’s tumor either under the skin (ectopic) or into the organ of tumor origin (orthotopic) and are most commonly used for preclinical drug development [102][103][104].
Physiologically, cells grow in three dimensions (3D) to form discrete tissue and organ structures [105][106]. PDO models, growing in 3D, have been shown to reliably recapitulate the architecture of the donor tissue and to preserve its genomic background, therefore providing a highly relevant physiological system [107]. With their optimal conditions for cellular proliferation, differentiation, and responsiveness to chemo- and targeted therapeutics, they recapitulate the functional tumor phenotype, including its ITH [41].
An additional advantage of PDO cultures is that they can be used in high-throughput drug screens. Such screens can be composed of multiple samples from the same tumor, thereby taking ITH into account [41][94].
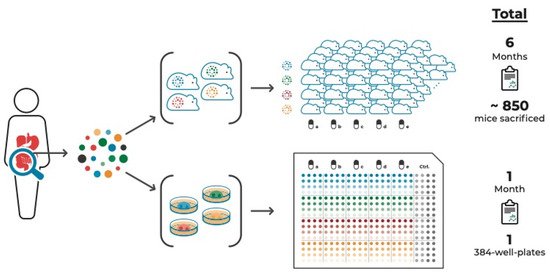
Figure 1.
It is known that the microenvironment with tumor-surrounding and infiltrating cells, including fibroblasts and immune cells, have a major impact on drug response [112][113]. Another significant advantage of the organoid model system is the ability to study the interaction of cancer organoids with other specific cell types that can be introduced into a direct or indirect co-culture system. Indirect co-culture systems are based on the use of cell conditioned media. They are simple to apply and are therefore often used for in vitro experiments [114]. However, they are not suitable for investigating the effects of cell contacts between cancer cells and stroma [115]. Direct co-culture models are a closer representation of the in vivo scenario. Since the tumor microenvironment plays a critical role in tumorigenesis, 3D co-culture systems are used, including not only cancer cells but also stromal cells [115]. Cancer cells were localized in a defined area within a stromal cell matrix to study the cytotoxic effect of anticancer drugs on both tumor and normal cells in the same system [116].
Since antibodies against immune checkpoint proteins/receptors have shown clear clinical benefit for patients with advanced cancer, including melanoma, non-small cell lung cancer (NSCLC), and mismatch repair deficient (dMMR) colorectal cancer, organoid co-culture systems including immune cells are moving into the spotlight of current in vitro application [117][118][119][120][121][122][123][124][125][126]. Dijkstra et al. established a co-culture system of autologous tumor organoids and peripheral blood lymphocytes of patients to induce and analyze tumor-specific T cell responses for mismatch repair deficient colorectal cancer and non-small cell lung cancer in a personalized manner [118]. Klein et al. demonstrated the advantages of co-culture systems of GBM organoids and human immune cells, to investigate not only immune–tumor interactions, but also to explore current and novel immunotherapies, such as adoptive T cell transfer, immune checkpoint inhibitors, or oncolytic viruses [127].
Another promising technology for studying ITH is Organ-on-a-Chip (OoC), a culture model to mimic complex and dynamic in vivo microenvironments [128]. An OoC is a multi-channel 3D microfluidic biochip, which recapitulates the activity, mechanism, and pathophysiological reaction of single-organ and multi-organ systems [129]. It is a useful tool in controlling spatial arrangement of cell growth and fluids within micrometer-sized channels, which may be used to increase the physiological relevance of tumor models [130]. OoC technology is expected to offer effective solutions to investigate the effects of drugs, as well as the causes of diseases and personalized therapeutic treatments [131][132][133].
The development of a multi-organoid platform that consists of patient-specific tumor organoids is currently in process. It is intended to offer the opportunity to test the efficiency of drug therapies designed based on genetic profiling. Skardal et al. generated a circulatory system with multiple tissue organoid sites by using microfluidic chip devices and used them to visualize and track tumor progression and kinetics of metastasis formation to distant site in vitro [132][133]. In combination with the even more complex body-on-a-chip platform, these personalized on-a-chip systems will be improved even further [132][133].