Primary aldosteronism (PA), a condition characterized by autonomous aldosterone hypersecretion, constitutes the most common cause of secondary hypertension. The advent and wide application of Next Generation Sequencing (NGS) technology led to the identification of several somatic and germline mutations associated with sporadic and familial forms of PA. Somatic mutations in ion-channel genes that participate in aldosterone biosynthesis, including KCNJ5, CACNA1D, ATP1A1, and ATP2B3, have been implicated in the development of aldosterone-producing adenomas (APAs). On the other hand, germline variants in CLCN2, KCNJ5, CACNA1H, and CACNA1D genes have been implicated in the pathogenesis of the familial forms of PA, FH-II, FH-III, and F-IV, as well as PA associated with seizures and neurological abnormalities.
- primary aldosteronism
- hypertension
- cardiovascular disease
- genetic causes of primary aldosteronism
1. Introduction
Hypertension is a major risk factor for the development of cardiovascular disease and its associated morbidity and mortality, and has been identified as the third leading cause of disability-adjusted life-years [1]. The global prevalence of hypertension for the year 2000 has been estimated to be 26.4% of the world population (976 million) and is expected to increase to 29% (1.56 billion) by 2025, with tremendous implications for the health care system [2].
Primary Aldosteronism (PA), a condition characterized by autonomous aldosterone overproduction, was first described by Conn in 1955 and was considered to be a rare cause of hypertension with a prevalence of less than 1% among hypertensive patients. However, following the introduction of plasma aldosterone concentration to plasma renin activity ratio (ARR) as a screening test in clinical practice, it became evident that the prevalence of PA is higher than previously thought [3][4][5][3,4,5]. Currently, PA is recognized as the most common cause of secondary hypertension, accounting for 6.1% of hypertensive patients and up to 20% of patients with severe or treatment-resistant hypertension [6][7][6,7]. Early identification and treatment of patients with PA is of paramount importance because they have a higher risk of stroke, myocardial infarction, and atrial fibrillation than patients with essential hypertension matched for age, gender, and blood pressure [8].
2. Evidence Favoring a Higher Prevalence of PA
The traditional approach in the diagnosis of PA involves an initial screening procedure to identify patients with higher probability for PA (Table 1) [9], followed by the application of aldosterone-to-renin ratio (ARR) test to detect elevated concentrations of aldosterone. Patients with positive ARR undergo a confirmatory test (e.g., oral-sodium loading, saline-infusion test, fludrocortisone suppression test, captopril test) in order to confirm or exclude autonomous aldosterone secretion independent of the renin-angiotensin system (RAAS), and subsequently an adrenal computed tomography (CT) scan to investigate the existence of an adrenal mass (bilateral or unilateral) [10]. Following this approach, Monticone et al. reported a prevalence of PA between 3.9% and 11.8%, depending on the severity of hypertension [11].
Table 1. The current recommendations of the Endocrine Society for Primary Aldosteronism screening.
The Current Recommendations of the Endocrine Society for Primary Aldosteronism Screening | ||
|---|---|---|
|
However, compelling evidence suggests that PA is much more prevalent than previously thought, with a large number of PA cases remaining undiagnosed [12][13][12,13]. This is mainly attributed to the fact that current diagnostic tests based on plasma aldosterone and renin concentrations may be insufficient to identify mild forms of PA, especially if based on the arbitrary conventional ARR thresholds [11]. Piaditis and others [13][14][13,14] have shown that when PA screening is extended to all hypertensive patients, irrespectively of the ARR, the PA prevalence is 3–5 times higher than previously determined, ranging between 16% and 31% across the spectrum of mild to severe hypertension [13][14][13,14]. Critically, PA has been documented in 11% and 13% of apparently healthy normotensive and normokalemic individuals that would escape diagnosis when the current guidelines are adopted or followed [13][14][13,14]. This becomes extremely prominent when considering the fact that 85% of a normotensive cohort with subclinical PA developed hypertension at 5-years of follow-up [15]. In line with these studies, administration of mineralocorticoid receptor antagonists (MRAs) has been shown to successfully control the blood pressure of patients with apparent essential hypertension, with their efficacy to be positively correlated with the levels of within the “normal” range aldosterone excess [16].
In light of these findings, Markou et al. investigated the role of ACTH, which has been predominantly regarded only as a minor regulator of aldosterone secretion in the development of PA. More specifically, they modified the standard fludrocortisone-saline suppression test (FST) by adding 2 mg of dexamethasone on the last day of testing (fludrocortisone suppression test, FDST) in order to eliminate the ACTH effect on aldosterone secretion [17]. Strikingly, after applying the FDST test to a cohort of hypertensive patients with normal ARR result and adrenal morphology, 31% of this cohort was diagnosed with PA and successfully treated [14]. In a subsequent study, we identified two novel germline mutations in the KCNJ5 gene in a cohort of patients without PA and normal adrenal CT, who exhibited ACTH-dependent aldosterone hypersecretion and responded to treatment with MRAs [18][19][18,19]. This work further expanded the clinical spectrum associated with KCNJ5 mutations and highlighted the importance of determining the genetic etiology of aldosterone hypersecretion in mild forms of primary aldosteronism towards earlier and targeted treatment.
3. Genetics of Primary Aldosteronism
PA can be either sporadic or familial. Sporadic cases of PA, Bilateral Adrenal Hyperplasia (BAH), and Aldosterone-Producing Adenomas (APAs), constitute the majority of PA cases (95%).
On the other hand, only 5% of cases are familial, and four familial forms of PA, FH-I, FH-II, FH-III, and FH-IV, which are inherited in an autosomal dominant manner, have been recognized to date [20].
4. Genetics of Sporadic Forms of Primary Aldosteronism
Prior to the advent of NGS, studies on sporadic forms of PA primarily focused on genetic variants influencing the susceptibility to PA or acting as phenotype modifiers, such as CYP11B2, a-Adducin, and Bradykinin B2 Receptor gene polymorphism [21]. The development and widespread use of NGS shifted our approach and led to the identification of several somatic mutations in APAs that not only identified APA-driver genes but also furthered our understanding of the molecular mechanisms of aldosterone production regulation [21]. The majority of the somatic mutations in sporadic forms of PA reported to date (Table 2) have been identified into well-characterized ion channels and ATPases genes involved in aldosterone biosynthesis pathway, including potassium channels (KCNJ5), calcium channels (CACNA1D, CACNA1H), and ATPases (ATP1A1, ATP2B3) [22]. More recently, somatic mutations in CTNNB1, PRKACA, and ARMC5 genes (Table 2) have been implicated in the pathogenesis of APAs [22].
Table 2. Mutations reported in sporadic primary aldosteronism.
Gene | Location (Hg19 *) | Somatic Mutations | Sporadic Germline Mutations | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
KCNJ5 | 11:128,761,251-128,790,930 | p.R115W | p.R52H | ||||||||
p.T126R | p.E145Q | ||||||||||
p.A139-F142dup | p.G151R | ||||||||||
p.F140L | p.G151E | ||||||||||
p.144-G145insAI | p.Y152C | ||||||||||
p.E145Q | p.I157S | ||||||||||
p.E145K | p.T158A | ||||||||||
p.E145_E147delinsK | p.G246K | ||||||||||
p.E147Q_T149_I150insTTT | p.E247R | ||||||||||
p.T148I | |||||||||||
p.T149insR | |||||||||||
p.T149S | |||||||||||
p.T149delinsTT | |||||||||||
p.T149delinsMA | |||||||||||
p.I150_G151insM | |||||||||||
p.G151R | |||||||||||
p.G153-G164dup | |||||||||||
p.F154C | |||||||||||
p.I157del | |||||||||||
p.I57_E159del | |||||||||||
p.I157K | |||||||||||
p.T158A | |||||||||||
p.L168R | |||||||||||
p.G184E | |||||||||||
p.R898W | |||||||||||
* Hg19: Human Reference Genome Hg19.
5. Genetics of Familial Forms of Primary Aldosteronism
Familial hyperaldosteronism is a rare cause of PA and is categorized into four types, each one characterized by specific clinical features and genetic causes (Figure 1) (Table 3). However, a considerable proportion of cases with early-onset PA remains unsolved, indicating that new genes responsible for FH remain to be identified [23][50].
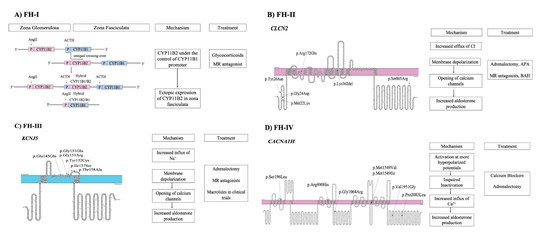
Figure 1. Pathogenetic mechanisms involved in familial forms of Primary Aldosteronism and treatment (A) FH-I is caused by an unequal crossover between CYP11B1 and CYP11B2 genes. (B) FH-II is caused by mutations in CLCN2 gene that lead to opening of calcium channels. (C) FH-III is caused by mutations in KCNJ5 gene. (D) FH-IV is caused by mutations in CACNA1H gene.
Table 3. Clinical characteristics of FH type I, II, III, and IV.
Familial Form of Primary Aldosteronism | Clinical Characteristics | ||||
|---|---|---|---|---|---|
Familial hyperaldosteronism type I, FH-I or glucocorticoid-remediable aldosteronism | Glucocorticoid-sensitive PATreated with dexamethasone administrationAdrenal hyperplasiaLow renin levelsIntracranial aneurysm and hemorrhagic strokeHypokalemiaNormotensive to severely hypertensiveHybrid steroid levels | ||||
Familial hyperaldosteronism type II | Phenotype resembling sporadic forms of PAHypokalemiaLow renin levelsGlucocorticoid-insensitive PAAPAs or Adrenal hyperplasia or bothNormotensive to severely hypertensive | ||||
Familial hyperaldosteronism type III | Low renin levelsBilateral massive adrenal hyperplasiaGlucocorticoid-insensitive PAHypokalemiaSeverely hypertensive | ||||
Familial hyperaldosteronism type IV | Low renin levelsNormal adrenal morphologyWide range of clinical phenotypes |
p.G246K | |||||||||
CACNA1D | |||||||||
3:53,529,076-53,846,492 | |||||||||
p.V259A | |||||||||
p.I770M | |||||||||
p.V309A | p.G403R | ||||||||
p.V401L | p.F747L | ||||||||
p.G403R | |||||||||
p.R619P | |||||||||
p.S652L | |||||||||
p.L655P | |||||||||
p.Y741C | |||||||||
p.F747L | |||||||||
p.F747C | |||||||||
p.F747V | |||||||||
p.V979N | |||||||||
p.P1336R | |||||||||
p.I750M | |||||||||
p.R990G | |||||||||
p.R993T | |||||||||
p.A998I | |||||||||
p.A998V | |||||||||
p.C1007R | |||||||||
p.I1015S | |||||||||
p.V1151F | |||||||||
p.I1152N | |||||||||
p.V1338M | |||||||||
p.V1353M | |||||||||
ATP1A1 | 1:116,915,795-116,947,396 | p.G99R | |||||||
p.F100L | |||||||||
p.P100_L104del) | |||||||||
p.L104R | |||||||||
p.V332G | |||||||||
p.I955_E960delinsK | |||||||||
p.960-963delp.A963S | |||||||||
ATP2B3 | X:152,801,580-152,848,387 | p.L425_V426del | |||||||
p.V426_V427del | |||||||||
p.V424_L425del | |||||||||
CTNNB1 | 3:41,240,942-41,281,939 | p.S33C | |||||||
p.G34R | |||||||||
p.A39Efs*3 | |||||||||
p.S45P | |||||||||
p.S45F | |||||||||
p.V426G_V427Q_A428_L433del | |||||||||
CLCN2 | 3:184,063,973-184,079,439 | p.Gly24Asp | |||||||
PRKACA | 19:14,202,507-14,228,559 | p.His88Asp | |||||||
p.Arg201Cys | |||||||||
p.Leu206Arg | |||||||||
ARMC5 | 16:31,470,317-31,478,488 | p.F14Y | |||||||
p.L156F | |||||||||
p.I170V | |||||||||
p.G323A | |||||||||
p.R502H | |||||||||
p.P507L | |||||||||
p.T643M | |||||||||
p.P826H | |||||||||
6. Familial Hyperaldosteronism Type I, FH-I
The first familial form of PA, described by Sutherland et al. [24][51] in 1966, was initially referred to as glucocorticoid-remediable aldosteronism (GRA) because it was treated with dexamethasone. However, it was renamed FH-I after the identification of additional forms of FH. The molecular basis of FH-I was delineated more than two decades after its first description, by the work of Lifton et al. [25][52], who recognized as its cause the presence of a chimeric gene produced by an unequal crossing-over event between the promoter region of 11-beta hydroxylase gene (CYP11B1) that participates in cortisol biosynthesis and the coding regions of aldosterone synthase (CYP11B2) gene. As a result, aldosterone is ectopically synthesized in the adrenal zona fasciculata under the regulation of ACTH and not under the renin-angiotensin system (RAS) [26][39]. The diagnosis of FH-I is based on genetic testing for the presence of the chimeric gene, while the treatment involves MRAs or glucocorticoid administration [7].