Inflammasomes are multiprotein complexes formed to regulate the maturation of pro-inflammatory caspases, in response to intracellular or extracellular stimulants. Accumulating studies showed that the inflammasomes are implicated in the pathogenesis of inflammatory bowel disease (IBD), although their activation is not a decisive factor for the development of IBD. Inflammasomes and related cytokines play an important role in the maintenance of gut immune homeostasis, while its overactivation might induce excess immune responses and consequently cause tissue damage in the gut. Emerging studies provide evidence that some genetic abnormalities might induce enhanced NLRP3 inflammasome activation and cause colitis. In these cases, the colonic inflammation can be ameliorated by blocking NLRP3 activation or its downstream cytokine IL-1β. A number of natural products were shown to play a role in preventing colon inflammation in various experimental colitis models. On the other hand, lack of inflammasome function also causes intestinal abnormalities.
- inflammasome
- inflammatory bowel disease
- NLRP3
- NLRP6
- Crohn’s disease
- ulcerative colitis
- IL-1β
- natural product
1. Introduction
1.1. Inflammatory Bowel Disease
Inflammatory bowel disease (IBD) is a chronic, relapsing disorder affecting the gastrointestinal (GI) tract [1][2][1,2]. IBD includes two main subtypes, Crohn’s disease (CD) and ulcerative colitis (UC). CD might affect any part of the GI tract but is most commonly found in the terminal ileum, cecum, peri-anal area, and the colon; it is marked by a dense inflammatory cell infiltration affecting all layers of the bowel wall. In contrast, inflammation in UC only affects the mucosal layer of the colon, causing infiltration of inflammatory cells [1]. IBD occurs in people of all ages with increasing incidence and prevalence worldwide [3]. Chronic colonic inflammation also increases the risk of developing colorectal cancer and, overall, causes serious social and economic burden [4][5][6][4,5,6].
Despite recent advances in our understanding of IBD, the exact pathogenesis of this disease remains elusive. A growing body of evidence supports the hypothesis that the onset of IBD is due to the interplay between host genetic, and environmental factors that cause an imbalanced mucosal homeostasis marked by an inappropriate immune response in the gut [7][8][9][7,8,9]. The innate immune system provides the first line of the host defense against pathogenic microbes and also plays an important role in regulating the immune homeostasis of the intestine [10][11][12][10,11,12]. As a major arm of innate immunity, the inflammasomes are considered to be prominent factors in modulating the development of inflammatory responses in the gut [13][14][13,14]. Mounting recent evidence shows that inflammasomes might have the potential to be a therapeutic target for treatment of IBD.
1.2. The Inflammasomes
Inflammasome is an intracellular multiprotein complex formed in response to the sensing of pathogen-associated molecular patterns (PAMPs) derived from invading pathogens, or danger-associated molecular patterns (DAMPs) derived from stress or injured cells. The concept of inflammasome was first proposed by Tschopp et al. in 2002 when they studied the function of NALP1 in regulating proinflammatory caspase activation and IL-1β maturation [15]. Canonical inflammasome assembly is initiated by the activation of an NALP (such as NLRP3 or AIM2) via its pattern recognition receptor, which then aggregates with an apoptosis-associated, speck-like protein containing a caspase recruitment domain (ASC) and caspase-1, to form a platform for caspase-1 activation [16][17][18][19][20][16,17,18,19,20]. In contrast, non-canonical inflammasome activation is initiated by cytosolic LPS or Gram-negative bacteria, which directly activates caspase-4 (caspase-11 in mice) and caspase-5. Activated caspase-1 induces the cleavage (i.e., maturation) of cytokine precursors (pro-IL-1β and pro-IL-18) as well as the cleavage of gasdermin D, which forms cell membrane pores that allow the release of the mature IL-1β and IL-18 from the cell. Gasdermin D is also acted on by caspase 4/5 (generated from the non-canonical inflammasome) and this also results in cleaved gasdermin D that mediates pore formation. Such a pore formation can lead to a form of cell death called pyroptosis [21][22][23][21,22,23].
To date, a number of inflammasomes are identified, including NLRP1, NLRP3, NLRC4, AIM2, and Pyrin [19][24][25][26][27][19,24,25,26,27]. Among these inflammasomes, the NLRP3 inflammasome is the most studied. Its activation requires two signals—the first signal (signal 1) consists of stimulation of membrane receptors such as TLRs and TNFR that prime NLRP3 and other inflammasome components by inducing their transcriptional expression and appropriate post-translational modification. The second signal (signal 2) consists of a variety of stimuli such as extracellular ATP or the ionophore nigericin that induce assembly of the inflammasome described above. The NLRP3 inflammasome can be activated by both PAMPs and DAMPs. Thus, it is not only implicated in host protective immune responses against invading pathogens, but also in the recognition of various endogenous danger signals that regulate the pathogenesis of a variety of human diseases, such as cancer and autoimmune diseases, including IBD [28][29][30][28,29,30].
1.3. The Inflammasomes Are Implicated in the Pathogenesis of IBD
Although the inflammasome processes IL-1β—a pro-inflammatory cytokine that is involved in the pathogenesis of many human diseases—it is not a decisive factor for ordinary IBD, since ordinary IBD patients do not respond to treatment by IL-1β blockade. In addition, patients with cryopyrin-associated periodic syndrome (CAPS) carrying a NLRP3 variant with a lowered activation threshold do not develop colonic symptoms indicative of IBD [31][32][33][31,32,33]. Nevertheless, previous studies showed that the production of IL-1β is increased in the serum of IBD patients and is associated with increased disease activity [34][35][34,35], indicating some level of involvement of inflammasomes and IL-1β in IBD. However, their precise contributions are controversial, at least for the NLRP3 inflammasome [36][37][38][39][36,37,38,39]. This is related to various factors, such as the genetic background of the mice or humans studied, the composition of the gut microbial community and the selection of the colitis model [28][40][28,40]. It is possible that inflammasome activation might contribute to the development of gut homeostasis in two ways. On one hand, activation that is regulated might play a protective role in the maintenance of gut immune balance; on the other hand, unregulated and excess activation might result in tissue damage and cause colitis [13][41][13,41].
2. NLRP3 Inflammasome Activation Contributes to Gut Homeostasis
Many recent studies showed that the NLRP3 inflammasome is involved in regulating colonic immune homeostasis. A good example of this is the findings reported by Huber et al. [42] showing that the NLRP3 and NLRP6 inflammasome-mediated production of IL-18 in the colon promotes the downregulation of IL-22 binding protein (IL-22BP), a soluble receptor expressed by immune cells that specifically binds to IL-22 and blocks the role of IL-22 in regulating colon epithelial cell repair. The epithelial repair function of IL-18 was also supported by studies of Zaki et al. [38], showing that mice with deficiency of NLRP3 inflammasome components exhibit increased Dextran Sodium Sulfate (DSS)-colitis and decreased epithelial integrity, as compared to the wild-type mice. Epithelial cells from these mice produced reduced levels of IL-18, and exogenous IL-18 reversed the increased colitis.
An NLRP3 inflammasome protective role also derives from studies using mice, carrying various genetic abnormalities affecting inflammasome regulators. For instance, Song et al. [43] showed that mice with absence of an E3 ubiquitin ligase, TRIM31, displayed enhanced NLRP3 inflammasome activation and reduced DSS-colitis. Similarly, Cui et al. [44] found that mice with macrophage-specific deficiency of CD1d1 also exhibited enhanced NLRP3 inflammasome activation and reduced DSS-colitis. The mechanism here might be that this deficiency enhances peroxiredoxin 1-associated ATK-STAT1 phosphorylation and subsequent NF-kB activation, and the latter causes increased transcription of NLRP3, IL-1β, and IL-18 in macrophages. Finally, abnormalities of protein tyrosine phosphatase non-receptor 22 (PTPN22) activity were shown to be inversely associated with colitis. Thus, Spalinger et al. [45] reported that PTPN22 deficiency causes pronounced colitis that is associated with inhibited inflammasome activation and IL-1β maturation due to increased NLRP3 phosphorylation at Tyr861. In contrast, an autoimmunity-associated PTPN22 variant (V619W) that leads to increased PTPN22 function causes increased NLRP3 inflammasome activation and excess IL-1β production that protects mice from experimental colitis. Thus, in these cases, upregulating the activity of the NLRP3 inflammasome might be a way to prevent the colonic inflammation.
3. Inhibition of the NLRP3 Inflammasome Ameliorates Colitis
The view that the NLRP3 inflammasome has a protective role in colitis, supported by the studies described above, is challenged by other studies showing that loss of this inflammasome function is associated with decreased colitis by one of several mechanisms, including decreased production of potentially damaging pro-inflammatory cytokines such as IL-18 or loss of suppressor cell development [46][47][46,47]. In addition, this view is inconsistent with a growing body of studies of various genetic abnormalities showing that excess inflammasome activation causes intensified colitis (summarized in Figure 1 and Table 1). In addition, the protective effect ascribed to NLRP3 inflammasome activation is not supported by many studies with animal models of colitis, which indicate that certain chemicals from natural products, microbes, probiotics, and stem-cell-based therapies prevent colonic inflammation by regulating the function of inflammasomes. Here, we summarize these studies and discuss their possible clinical applications to IBD.
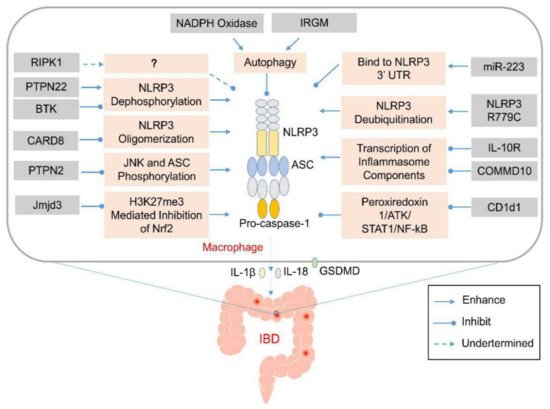
Figure 1. Genetic abnormalities affect development of inflammatory bowel disease (IBD) by elevating the activation of inflammasomes via various cellular processes. Various genetic factors might prevent the activation of the NLRP3 inflammasome via various mechanisms that affect NLRP3 oligomerization or modulate autophagy. Their absence might induce aberrant activation of the NLRP3 inflammasome and excess production of IL-1β and IL-18 in the infiltrating macrophages or dendritic cells in the lamina propria of the colon. The released pro-inflammatory cytokines and pyroptosis-mediated release of cellular contents might recruit other inflammatory mediators to promote the expansion of the colonic inflammation.
Table 1. Genetic factors affect IBD by regulating the activation of inflammasomes.
Gene | Function to Inflammasome | Mechanism | Deficiency or Variant | Variant Impact to Disease | Disease or Model | Reference | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
IRGM | Inhibit NLRP3 inflammasome assambly | Interact with NLRP3 and Promote NLRP3 autophagic degradation | Deficiency | Exacerbate colitis | DSS-colitis in mouse |
[48] |
[49] |
|||||||||||||
RIPK1 | Inhibit NLRP3 inflammasome activation upon LPS stimulation | Not clear | Deficiency | Primary immunodeficiency and/or colitis | Human colitis |
[49] |
[51] |
|||||||||||||
BTK | Inhibit NLRP3 inflammasome activation | Inhibit PP2A mediated NLRP3 dephosphorylation | Deficiency | Exacerbate colitis | DSS-colitis, TNBS-colitis in mouse |
[50] |
[53] |
|||||||||||||
PTPN2 | Inhibit NLRP3 inflammasome activation | Inhibit JNK and ASC phosphorylation | Deficiency in myeloid cell | Exacerbate colitis | DSS-colitis in mouse |
[51] |
[54] |
|||||||||||||
Jmjd3 | Enhance NLRP3 inflammasome activation | Prevent H3K27me3 mediated inhibition of Nrf2 | Inhibition or Knock-down | Ameliorate colitis | DSS-colitis |
[52] |
[55] |
|||||||||||||
miR-223 | Inhibit NLRP3 inflammasome activation | Bind to NLRP3 3’ untranslated region to inhibit inflammasome assembly | Deficiency | Exacerbate colitis | DSS-colitis in mouse |
[53] |
[56] |
|||||||||||||
NLRP3 mutation | Enhance NLRP3 inflammasome activation | Enhance deubiquitination of NLRP3 via binding with BRCC3 and JOSD2 | R779C | Exacerbate colitis | DSS-colitis in mouse |
[54] |
[61] |
|||||||||||||
COMMD10 | Inhibit NLRP3 inflammasome activation | Inhibit transcription of inflammasome components | Deficiency in monocytes | Exacerbate colitis | DSS-colitis in mouse |
[55] |
[62] |
|||||||||||||
CARD8 | Inhibit NLRP3 inflammasome assembly | Interact with NLRP3 and inhibit NLRP3 oligomerization | V44I | Exacerbate colitis | Crohn’s disease |
[56] |
[63] |
|||||||||||||
IL-10R | Inhibit NLRP3 inflammasome activation | Inhibit expression of NLRP3 and IL-1β | Deficiency | Spontaneous colitis in mouse and infant-onset IBD in human | Mouse spontaneous colitis and human IBD |
[57] |
[64] |
|||||||||||||
NADPH oxidase | Inhibit NLRP3 inflammasome activation | Induce autophagy | Deficiency | Exacerbate colitis | TNBS-colitis in mouse |
[58] |
[65] |
|||||||||||||
CD1d1 | Reduce transcription of NLRP3 inflammasome components | Reduce peroxiredoxin 1/ATK/STAT1 mediated NF-kB signaling | Deficiency in macrophage | Ameliorate colitis | DSS-colitis in mouse |
[44] |
||||||||||||||
PTPN22 | Enhance NLRP3 inflammasome activation | Induce NLRP3 dephosphorylation at Tyr861 | Deficiency | Exacerbate colitis | DSS-colitis in mouse |
[45] |
||||||||||||||
V619W | Ameliorate colitis | DSS-colitis in mouse |
[45] |
Abbreviations: IRGM: Immunity-related GTPase family M; RIPK1: Receptor interacting protein kinase 1; BTK: Bruton’s tyrosine kinase; XLA: X-linked agammaglobulinemia; PTPN2, 22: protein tyrosine phosphatase non-receptor type 2, 22; Jmjd3: Jumonji domain-containing 3; VEOIBD: Very-Early-Onset inflammatory bowel disease; COMMD10: COMM domain containing 10; CARD8: caspase recruitment domain family member 8; CGD: chronic granulomatous disease; PP2A: serine/threonine protein phosphatase 2A; DSS: Dextran Sodium Sulfate; TNBS: 2,4,6-trinitrobenzene sulfonic acid; PPARγ: peroxisome proliferator-activated receptor γ; SO2: sulfur dioxide; and CysA: cyclosporin A.
4. Regulation of Inflammasomes Other Than NLRP3 in Colitis
4.1. The NLRP6 Inflammasome
The NLRP6 inflammasome is an important player in the maintenance of intestinal homeostasis. Elinav et al. [59][103] studied the role of NLRP6 in colitis, and showed that NLRP6-deficient mice exhibited exacerbation of DSS-induced colitis, due to the induction of colitogenic microbiota. Using co-housing experiments, they found that the colitogenic microbiota were transferable and can cause intensified colitis in wild-type mice. Following this line, the authors in further studies found that some metabolites generated by the microbiota were able to regulate the host-microbiota interface by modulating NLRP6, IL-18, and anti-microbial peptides in the epithelia. The colitogenic microbiota might regulate AMP balance to favor its own colonization via metabolite-mediated inflammasome inhibition [60][104]. Thus, restoration of the metabolite–inflammasome–AMP axis might be used in disease intervention.
The susceptibility of NLRP6-deficient mice to chemically-induced colitis was confirmed by Chen et al. [61][105]. These authors showed that loss of NLRP6 was associated with impaired production of IL-18 and increased epithelial injury. In further studies they found that NLRP6 plays a role in inhibiting the spontaneous colitis in IL-10 deficient mice, probably by inducing the enrichment of Akkermansia muciniphila, a bacterium that can promote development of colitis [62][106].
For the regulation of the NLRP6 inflammasome, Mukherjee et al. [63][107] found that the assembly of this inflammasome was regulated by deubiquitinase Cyld, which mediates deubiquitination of NLRP6; as a consequence, Cyld inhibits NLRP6-ASC assembly and IL-18 production. Cyld deficiency causes elevated levels of mature IL-18 and severe colonic inflammation following Citrobacter rodentium infection.
4.2. The NLRP1 Inflammasome
A number of studies investigated the role of NLRP1 in pathogenesis of IBD. The first such study was conducted by Williams et al. [64][108] who showed that Nlrp1b-deficient mice exhibited intensified colitis induced by DSS as compared to wild-type mice. The increased inflammation in Nlrp1b-deficient mice was correlated with reduced production of IL-1β and IL-18 in nonhematopoietic cells in the colon. However, these results were inconsistent with those reported by Tye et al. [65][109], showing that Nlrp1 deficiency suppressed DSS-induced colitis by promoting the expansion of beneficial, butyrate-producing Clostridiales. Moreover, an activating mutation of NLRP1 aggravated DSS-induced colitis, which was associated with an enhanced Th1 response and an increased IL-18/ IFNγ production in the gut. Further studies demonstrated that the colitis in mice carrying activated Nlrp1 was dependent on IL-18. The expression of NLRP1, IL-18, or IFN-γ, negatively correlated with the abundance of Clostridiales in biopsies from the patients with ulcerative colitis. Therefore, the contribution of NLRP1 to colonic inflammation might be associated with the composition of gut microbiota, especially the butyrate-producing Clostridiales found in the Tye et al. study.
4.3. The Pyrin Inflammasome
Mutations in the pyrin gene (Mefv) is associated with hereditary autoinflammatory disease and IBD. In a study by Sharma et al. [66][110] it was shown that deficiency of Mefv caused increased DSS-colitis with increased epithelial permeability and loss of tight junction proteins. These manifestations were associated with reduced production of IL-18 in the colon, because administration of recombinant IL-18 increased tight junction protein expression, reduced epithelial permeability, and thus reduced the intensified inflammation of Mefv-deficient mice. Thus, compensation of IL-18 might work as a therapy for patients with Mefv mutation or deficiency, but further clinical studies are required to clarify this issue.
4.4. The AIM2 Inflammasome
Ratsimandresy et al. [67][111] investigated the role of the AIM2 inflammasome in IBD. They showed that AIM2 deficiency causes loss of IL-18 and IL-22BP expression in intestinal epithelial cells, which was followed by the loss of STAT3-mediated expression of antimicrobial peptides. In the DSS-induced colitis model, AIM2 deficiency induced excessive production of IL-22, enhanced activation of STAT3 and Akt, and promoted the expression of AMPs Reg3b and Reg3g. Thus, the AIM2 inflammasome prevents intestinal inflammation by regulating the IL-18/IL-22BP/IL22/STAT3 pathway and AMP expression.
4.5. The NLRC4 Inflammasome
NLRC4 is an intracellular flagellin receptor. Carvalho et al. [68][112] assessed the role of NLRC4 in the development of colonic inflammation. Their data showed that NLRC4 deficiency did not alter the gene expression profile induced by the administration of flagellin. However, NLRC4-deficient mice manifested intensified colitis induced by DSS compared with wild-type mice. In further studies, the investigators showed that NLRC4 deficiency caused increased mortality after infection with Salmonella. Thus, NLRC4 inflammasome-mediated production of IL-1β and IL-18 might be the key factors protecting the host from colonic inflammation and infection.