There is an emerging role of the transcriptional regulator WW domain containing oxidoreductase (WWOX) in a number of neurological disorders including early-onset epilepsy, autism spectrum disorder (ASD), multiple sclerosis (MS), and Alzheimer’s disease. However, the underlying mechanism of WWOX function is still currently unknown.
- WOREE
- DEE28
- SCAR12
- WWOX
- epilepsy
1. WWOX in Alzheimer’s Disease
1. WWOX in Alzheimer’s Disease
Mutations in WWOX have recently been found to be a significant risk factor for disease development in Alzheimer’s disease, an adult-onset neurodegenerative disorder [1]. Furthermore, decreased WWOX protein levels have been observed in Alzheimer’s disease patients compared to age-matched healthy controls [2][3]. In the hippocampus of Alzheimer’s disease patients, an increase in the phosphorylation of the microtubule-associated protein, tau, was reported in a glycogen synthase kinase 3β (GSK-3β)-dependent pathway [3]. WWOX was found to physically interact with and inhibit GSK-3β, preventing tau hyperphosphorylation and transformation into neurofibrillary tangles (NFTs), leading to neuronal loss [4]. WWOX was also shown to promote neural differentiation by suppressing GSK-3β activity, resulting in an increase of the affinity to microtubules of tau, allowing microtubule assembly to prompt neuronal differentiation [4] (
Figure 1). It has also been demonstrated that WWOX physically interacts with TPC6AΔ, an aggregated vesicle-trafficking protein isoform that has a critical role in causing caspase activation, tau aggregation, and Aβ generation in patients with Alzheimer’s disease, blocking its self-aggregation [5]. Consequent to WWOX loss, TPC6AΔ undergoes polymerization leading to the aggregation of TGF-β1-induced antiapoptotic factor (TIAF1) caspase activation, which causes amyloid precursor protein (APP) degradation, leading to the generation of amyloid β and the formation of the neurofibrillary tangles (NFTs), causing neurodegeneration (
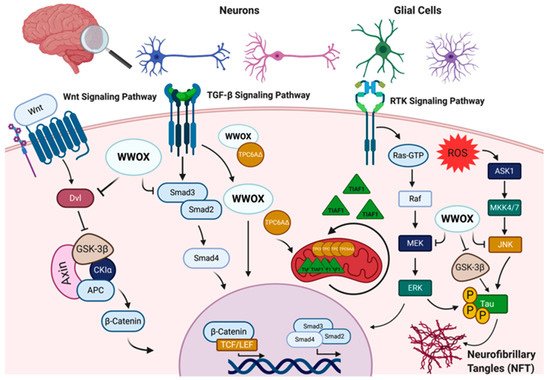
Figure 1. WW domain-containing oxidoreductase (WWOX) interaction partners and signaling pathways in the nervous system. WWOX protein–protein interactions and signaling in the nervous system involves: I. The Wnt/β-catenin signaling pathway, where WWOX negatively regulates this pathway through interaction with Dishevelled (Dvl) proteins and/or glycogen synthase kinase-3 β (GSK-3β). WWOX deficiency has been shown to be associated with increased nuclear β-catenin activity. II. WWOX has been also shown to modulate the transforming growth factor-β (TGF-β) signaling pathway through regulation of the SMAD proteins and the trafficking protein particle complex 6AΔ (TPC6AΔ). In the absence of WWOX, the SMAD/co-SMAD complex is inhibited, and TPC6AΔ and anti-apoptotic factor 1 (TIAF1) aggregate in the mitochondria, contributing to CNS pathologies. III. WWOX could also modulate the receptor tyrosine kinase (RTK) and c-Jun N-terminal kinase (JNK) signaling pathways to regulate tau phosphorylation and the formation of neurofibrillary tangles (NFT). Other abbreviations used: APC, adenomatosis polyposis; CKIα, casein kinase Iα; TCF/LEF, T-cell factor/lymphoid enhancing factor; ROS, reactive oxygen species; ASK1, apoptosis signal-regulating kinase 1; MKK4/7, mitogen-activated kinase kinase 4/7; RAF, rapidly accelerated fibrosarcoma; MEK: mitogen-activated protein Kinase; ERK, extracellular signal-regulated kinase. Figure was created via BioRender.
2. WWOX in Multiple Sclerosis
Multiple sclerosis (MS) is an immune-mediated disorder that affects the CNS [8]. It is a demyelinating disease that attacks the myelin sheaths covering the axons of nerve cells in the brain and the spinal cord. These attacks cause myelin sheath destruction, which can further lead to axonal damage and loss, inducing many severe neurological disabilities [9]. Lately, a study published by Jäkel et al. using single nucleus RNA-sequencing on post-mortem tissues of 4 MS individuals and 5 healthy controls determined that an alteration in oligodendrocyte population heterogeneity between MS individuals and the controls and a shift in the transcription profile of the different oligodendrocyte sub-clusters in MS tissues. Markedly, in their differential gene expression analysis, WWOX levels were shown to be reduced in the different types of MS lesions, mostly in the chronic active lesions, compared to the controls [10].
2. WWOX in Multiple Sclerosis
p =
−11 and an odds ratio (OR) of 1.08 [11]. WWOX rs7201683, another variant, was shown to have a higher frequency in MS individuals than in healthy controls in a study performed on 3 Italian families with MS members, 120 unrelated MS individuals, and healthy controls in order to identify new exonic low-allele frequency variants associated with MS [12]. Furthermore, in a GWAS on 776 MS individuals and 75 healthy controls conducted to study the gray matter pathology indicated by cortical thinning in MS, WWOX was noted to be associated with the thinning of different cortical regions in MS [13]. Additionally, in an analysis of genetic data from 15 different MS genome-associated studies of 47,429 MS individuals and 68,374 healthy controls, the rs12925972 WWOX variant was suggested to be significantly associated with MS susceptibility with a
p =
−8 and an OR of 1.099 [14]. Altogether, WWOX could play important roles in MS, perhaps due to its emerging function controlling myelination and oligodendrocyte differentiation.
3. WWOX in Autism Spectrum Disorders (ASD)
Heterozygous deletions and duplications overlapping the WWOX gene have been observed in individuals with ASD [15][16]. Copy number variants (CNVs) of WWOX have been reported in many ASD individuals characterized with less severe ASD phenotypes and intelligence quotient (IQ) levels approximate to the normal ranges, suggesting that heterozygous WWOX variants were a low-penetrance risk factor for ASD [17][18]. These WWOX variants act as weak risk factors and are generally associated with milder ASD phenotypes.
4. WWOX in Early Onset Epilepsy and Ataxia
Consistent with the observations in WWOX animal models, human patients with WWOX bi-allelic loss of function variants (deletions, nonsense, and some missense mutations) were found to be associated with autosomal recessive cerebellar ataxia, epilepsy, optic tract atrophy, retinal degeneration, growth retardation, developmental delay, intellectual disabilities, microcephaly with seizures, and early death [19][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38]. The range of phenotypic severity of WWOX-deficient individuals extends from less severe phenotypes with later onset and non-progressive microcephaly, as seen in SCAR12, to more severe phenotypes with progressive microcephaly, seizures, global developmental delay, optic atrophy, and early age lethality, as observed in WOREE. Moreover, brain magnetic resonance images (MRI) of children with WOREE revealed an abnormally thin or hypoplastic corpus callosum, progressive optic atrophy, and brain atrophy as the most common signatures, while delayed myelination and white matter hyperintense signals were reported in some cases [20]. In patients with SCAR12, brain MRIs revealed mild hypoplasia of the cerebellum or cerebellar vermis. There was also a description of a patient with epilepsy of infancy with migrating focal seizures (EIMFS), in which the seizures started at 2.5 months with infantile spasms, and at the age of 6 years, there was profound severe intellectual impairment, microcephaly, spasticity, hypotonia, and scoliosis [35]. Another interesting West syndrome case described a heterozygous 6.8-Mb deletion of WWOX suggesting other genes within this large genomic region might be involved [39]. Although there are limited studies, overall, the phenotypic severity of WWOX deficient patients appears to be variant-specific, suggesting a phenotype–genotype correlation.