The c-Jun N-terminal kinase (JNK) signalling pathway is a conserved response to a wide range of internal and external cellular stress signals.
- MAP kinase
- cell signalling
- glioblastoma
- neurodegenerative disorders
- CNS injuries
1. Introduction
The CNS is exposed to stress stimuli during development and adulthood, and under pathological aggressions. These events trigger the Jun N-terminal kinase signalling pathway (JNK) as a mechanism to coordinate cellular responses to stress, and maintain tissue homeostasis. The JNK pathway includes a conserved mitogen-activated protein kinase (MAPK), which belongs to the stress-activated protein kinase (SAPK) group, a group of kinases that can be activated by any internal or external stimuli that cause cell stress. Furthermore, the JNK pathway in the CNS and other tissues can be activated by UV irradiation, glucose deprivation, DNA damage, heat stress, bacterial and viral infection, oxidative stress, inflammatory cytokines and growth factors [1][2][3][4][5][6][7][8][9][10][1,2,3,4,5,6,7,8,9,10].
The molecular MAPK cascade presents high homology from Drosophila to mammals (Figure 1) [11]. In Drosophila, JNK pathway-signalling is initiated by the interaction of the ligand protein Eiger (Egr), the unique TNF superfamily member of ligands [12][13][12,13], with TNF receptors (TNFRs) Grindewal (Grnd) [14], or Wengen (Wng) [15]. Ligand–receptor interaction initiates a cascade of phosphorylations that mediates the JNK signalling pathway [11]. In Drosophila, JNK is encoded by a single gene: basket (Bsk); this simplifies the genetic studies that have contributed to decipher the role of JNK under physiological and pathological stressful scenarios [16][17][16,17]. In mammals, the JNK cascade involves four kinases, and mitogens or cytokines induce MAP3K family activation [11]. MAPK cascade triggers cytosolic JNK dual phosphorylation and initiates the phosphorylation of cytoplasmic and nuclear proteins [18], including cytoskeletal and mitochondrial proteins, nuclear transcription factors, membrane proteins or nuclear hormone receptors [19]. Thus, gene expression derived from JNK activation leads to a variety of responses, depending on the cell type and the scenario [11]. In mammals, the JNK pathway is encoded by jnk1, jnk2 and jnk3 [20]. JNK1 and JNK2 are found ubiquitously, whereas JNK3 is restricted to the brain, cardiac smooth muscle and testis [20][21][20,21]. In particular, the JNK pathway is highly active in the CNS as compared with other tissues. Therefore, it emerges as a critical regulator of CNS cells under physiological and pathological conditions [22][23][22,23]. It has been demonstrated in mouse that different JNK isoforms undergo compensatory mechanisms during early brain development. Although a single deficiency of each JNK isoform is viable, the mutants still present different phenotypes, highlighting the importance of each individual enzyme (reviewed in [23]). Besides, each isoform has different temporal and regional expression patterns (reviewed in [21]).
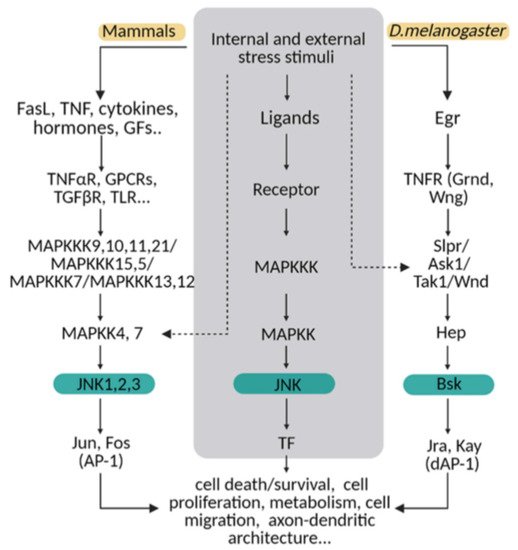
Figure 1. The JNK pathway is conserved between mammals and flies. Upon internal and external stress stimuli, ligands (ex. TNF/Egr) activate transmembrane receptors (ex. TNFαR/ Grnd) and initiate a cellular response based on a MAPK cascade phosphorylation. In mammals, different ligands can trigger the signal path; while in Drosophila, Eiger (Egr) is the TNF ligand that predominantly initiates the stress response via TNF receptors (Grindewal, Grnd; or Wengen, Wng). In mammals, there is MAPK path gene redundancy that is shown in this diagram compared with flies. MAPKs are encoded by multiple genes in mammals, whereas in Drosophila, one single gene is described for each enzyme. The cellular response converges in JNK/Bsk phosphorylation, which triggers the expression of transcription factors as Jun/Jra and Fos/Jay, known as an AP-1/dAP-1 complex. Finally, AP1/dAP-1 modulates the transcriptional program of genes involved in a variety of biological activities. This pathway can be activated at other steps indicated with dotted lines in the diagram.
The JNK signalling pathway mediates embryonic development; metabolism and growth; lifespan; programmed cell death; cell migration, repair and proliferation; immunity; and axonal transport [24][25][26][27][28][24,25,26,27,28]. In addition, the literature reveals a dual role of the JNK pathway in cell death and cell survival, depending on the cell type and the context [11]. This two-faded role is particularly important in CNS pathologies such as neurodegeneration and tumorigenesis, in which the cellular stress-associated signals are increased [29][30][29,30]. Likewise, JNK signalling is involved in neurogenesis, axonal growth, axonal transport, brain metabolism, animal behavior, neurulation, migration and axon–dendritic architecture in different species [21][22][23][31][32][21,22,23,31,32].
2. JNK in Regeneration/Repair after an Injury to the CNS
CNS damage produced by brain stroke, spinal-cord injury or neurodegenerative diseases often results in permanent disabilities due to the limited regenerative capacity of this tissue. Several studies have supported the importance of JNK signalling in response to nerve injuries in both degeneration and repair. In the injured axons, retrograde transport of JNK can alter somal transcription of the injury-response molecules ATF3 and Hsp27, important for axonal outgrowth [33][34][72,73]. On the other hand, genetic or pharmacological inhibition of JNK signalling in multiple models of axonal injury delayed axonal degeneration [35][36][74,75], indicating that JNK is also required for axonal degeneration. Moreover, upon nerve injury of a rat model, JNK3 inhibited axonal growth through the interaction with the Kluppel-like transcription factor 9 (KLF9) [37][76]. JNK signalling also activates members of the Bcl-2 family, triggering apoptosis after injury or stress [38][39][40][77,78,79].
MAP3K is one of the first members of the JNK pathway. It is a dual-leucine zipper kinase (DLK), a conserved kinase with orthologues in mammals (MAP3K DLK), Drosophila (DLK/Wallenda) and C. elegans (DLK-1) [41][80]. MAP3K is an important axon-injury sensor [42][81]. DLK–JNK signalling contributes in multiple ways to axonal injury response and axonal regeneration. The DLK/Wallenda protein is present in axons, and protein levels are increased in response to axonal injury in Drosophila [43][44][82,83]. JNK-dependent phosphorylation of DLK is required for the stabilization of DLK levels [45][84]. DLK regulates microtubule stability [42][46][47][81,85,86], essential for axonal regeneration in spinal-cord injury [48][87]. DLK/Wallenda overexpression has also been shown to be protective in Drosophila motor neuron axons [44][83]. DLK is also an essential molecule for the injury-dependent activation of the retrograde transport of p-STAT3 to the cell body, necessary for the activation of the neuronal regenerative program in mice [49][88]. Moreover, it has been demonstrated that in C. elegans, DLK-1 is both necessary and sufficient for injury-induced autophagy activation; DLK-1 limits the levels of LIN-12 and NOTCH proteins, suggested to promote axon regeneration [50][89].
2.1. JNK Signalling in Glial Cells upon Injury
All the evidence summarized above demonstrates the importance of JNK signalling in injured axons; however, JNK-signalling activation also occurs in glial cells in response to injury. Different injury paradigms in Drosophila showed a consistent activation of AP-1 transcription in glial cells upon injury, downstream of JNK. Unknown molecules from axonal debris activate the engulfment receptor Draper, which in turn regulates draper transcriptional upregulation via STAT92E [51][90] and the JNK pathway [35][74]. Draper is a conserved receptor with orthologues in mammals (MEGF10) and C. elegans (CED-1), and it is required for engulfment in several cell types, including germ line, epithelial cells, microglia and astrocytes [52][53][54][55][91,92,93,94].
In Drosophila and C. elegans, downregulation of JNK reduces axon regeneration and provokes axon debris accumulation following injury. In the injury context, JNK also activates MMP-1 expression [56][95], which is required for glial cells to infiltrate in the injured tissue and remove cell debris via Draper. Glial-specific overexpression of draper is sufficient to rescue engulfment defects associated with the loss of JNK signalling [35][74]. In addition, knockdown of the JNK pathway components blocks CED-1 mediated axon regeneration and axon debris removal [57][96], which suggests a role for JNK in Drpr/CED-1-mediated axon regrowth.
2.2. JNK in Neurogenesis and Regeneration
Finally, JNK could also trigger CNS regeneration by promoting neuronal differentiation from neural stem cells (NSCs). So far, neurogenesis in mammals is restricted to two major regions, the subependymal zone (SEZ) and the subgranular zone (SGZ) of the dentate gyrus, where adult NSCs are present, but the neuronal differentiation rate of NSCs is limited [58][97]. Therefore, a promising strategy to boost CNS regeneration is to find candidates to promote neural differentiation from NCSs. Noncanonical Wnt signalling such as the Wnt/JNK pathway has a positive effect on neuronal differentiation in cell culture and during development [59][60][61][98,99,100]. Moreover, it has been recently shown that Wnt5a upregulates miRNA200b-3p expression through MAPK/JNK signalling, and miRNA200b-3p suppresses the RhoA/Rock signalling required for neuronal differentiation. Thus, Wnt5a promotes NSC differentiation into neurons, and more remarkably, transplantation of NSCs overexpressing Wnt5a results in tissue repair and locomotor functional recovery in rats after spinal-cord injury [58][97]. However, other studies have revealed that different JNK isoforms are involved in the adult neurogenesis control in a different manner. For instance, inhibition of JNK1 or mice lacking JNK1 show an increased number of neural progenitors in the SGZ of the hippocampus, whereas the absence of JNK3 reduces it [62][63][101,102]. These results again point out the importance of finding drugs with high specificity for the different JNK isoforms to treat CNS disorders.
(References would be added automatically after the entry is online)