The crucial role played by compounds bearing amide functions, not only in biological processes but also in several fields of chemistry, life polymers and material sciences, has brought about many significant discoveries and innovative approaches for their chemical synthesis. Within this context, and following the development of C-H functionalization strategies, C-H amidation reactions have only recently emerged as valuable approaches for the construction of amide functions. These methodologies have been successfully used for both C(sp2)- and C(sp3)-H activations and blossoming synthetic applications have appeared in the last two years (2019-2020 timeframe).
- amides
- C-H functionalization
- enantioselectivity
- dioxazolones
- metal-transition catalysis
1. Introduction
Amides are ubiquitous and one of the most important functional groups in organic, medicinal, coordination and natural products chemistries, and in the fields of polymers, material and life sciences [1,2][1][2]. If several efforts have been devoted to finding new practical synthetic methods to allow their preparation through less conventional ways (i.e., avoiding amine-carboxylic acid couplings with activating agents) [3[3][4],4], the development of more “direct” strategies is still underdeveloped. Within this context, and following the development of C-H functionalization strategies, C-H amidation reactions have only recently emerged as valuable approaches for the construction of amide functions (Scheme 1). These methodologies have been successfully used for both C(sp2)- and C(sp3)-H activations and blossoming synthetic applications have appeared in the last few years.
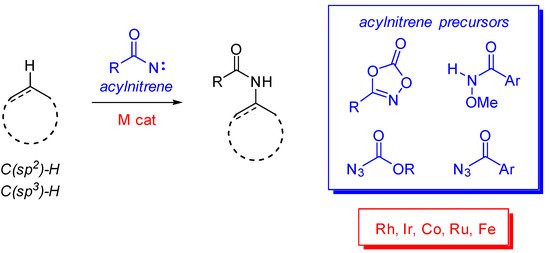
Scheme 1. C-H amidation reactions.
The reasons for such tremendous developments rely on the following several key points:
A major breakthrough in C-H amidation reactions was the introduction by Chang in 2015 of dioxazolones 1 as acylnitrene precursors [5]. Dioxazolones are thermally stable and are easily obtained from the corresponding hydroxamic acid under green, mild and scalable conditions (Scheme 2) [6,7][6][7]. The acylnitrene transfer usually occurs under mild conditions, at room temperature or 40 °C, in the absence of any stoichiometric external oxidant [8,9][8][9]. By the way, to the best of our knowledge, all the enantioselective C-H amidations reported to date involve the use of dioxazolone as acylnitrene precursors.
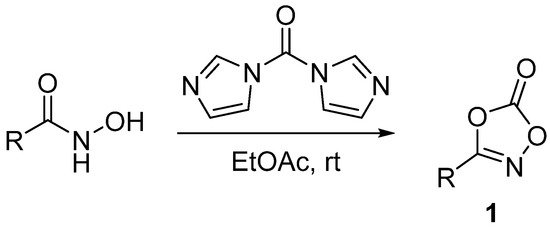
Scheme 2. Dioxazolones 1 synthesis.
If C-H amidation reactions were initially carried out in the presence of noble and expensive metals (Rh and Ir), the use of abundant, less toxic and cheap first row metal cobalt has been also developed [10]. Very recently, the use of (Phthalocyanine)FeIIICl under aerobic conditions was also described in intramolecular C(sp3)-H amidations with remarkable high turnovers [11]. A simplified mechanistic manifold for C(sp2)-H amidation is illustrated in Figure Figure 11, but significant differences exist depending on the metal used [7,11,12][7][11][12].
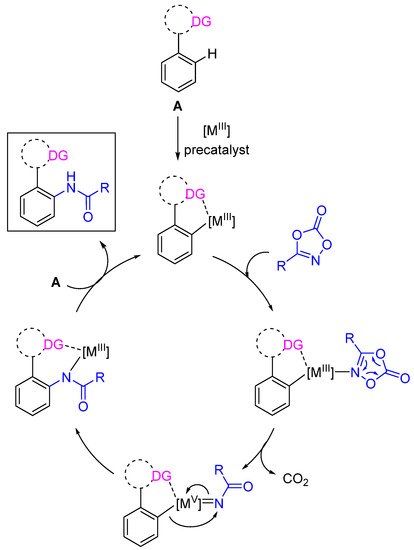
Figure 1. Simplified mechanism for C(sp2)-H amidation. DG = Directing Group.
In C(sp2)-H directed functionalizations, C-H amidations constitute interesting alternatives to Buchwald-Hartwig, Ullmann or Chan-Lam strategies particularly within the context of medicinal chemistry in which amide bonds associated with N-heterocyclic platforms are prevalent in drug candidates [13,14][13][14]. As a matter of illustration, Scheme 3 highlights a non-exhaustive collection of C-H amidation products described in the last two years [15,16,17,18,19,20,21,22,23,24][15][16][17][18][19][20][21][22][23][24].
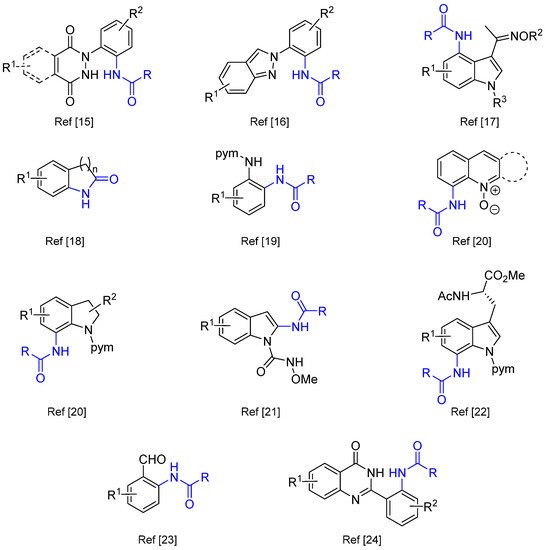
Scheme 3. Recent (non-exhaustive) examples of products issued from Csp2-H amidation reactions. pym = pyrimidyl.
The potential of C(sp2)-H functionalizations has been brilliantly illustrated by Ellman and Miller in the structural diversification of thiostrepton, a potent antibiotic peptide leading to analogs with maintained biological activities while increasing aqueous solubility (up to 28-fold) (Scheme 4) [25]. Thiostrepton presents three dehydroalanine (Dha) residues. A regioselective Co(III)-catalyzed C(sp2)-H amidation of a single Dha moiety provided an elegant entry to novel analogs with increased physicochemical properties. Undoubtedly, this work opens the door to future achievements on late-stage functionalization of other natural products.
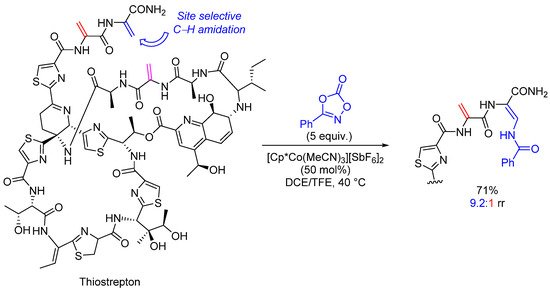
Scheme 4.
C(sp
2
)-H functionalization for the structural diversification of thiostrepton.
Besides, intramolecular C(sp3)-H functionalizations, branch-selective allylic C-H amidation of terminal double bonds have been described [26,27,28,29][26][27][28][29]. Based on stoichiometric studies with TsNH2 and the isolation of an allyl-Ir(III) complex, an inner sphere nitrenoid insertion of an η3-allyl irididium intermediate is advocated in these reactions (Scheme 5) [26].
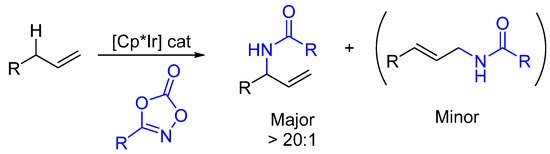
Scheme 5. Regio(branch)-selective allylic C(sp3)-H amidation.
Thus, these notable pivotal achievements have set the stage to future advances on the field of C-H amidation reactions. Indeed, a new benchmark has now been achieved with the development of enantioselective C-H amidation reactions in both C(sp3)-H and C(sp2)-H functionalizations with the publication of about ten papers in the 2019–2020 period. This review intends to give to readers an overview of very recent (last two years) applications of enantioselective C-H functionalization for amidation reactions.
2. Enantioselective C(sp3)-H Amidations
Early 2019, Matsunaga and Yoshino have described the first example of an intermolecular C(sp3)-H enantioselective amidation of thioamides [49,50][30][31]. In the presence of an achiral Co(III) catalyst, and a bulky chiral carboxylic acid derived from tert-Leucine, β-amino thiocarbonyl derivatives bearing an α-quaternary center were obtained in high enantioselectivities (Scheme 6).
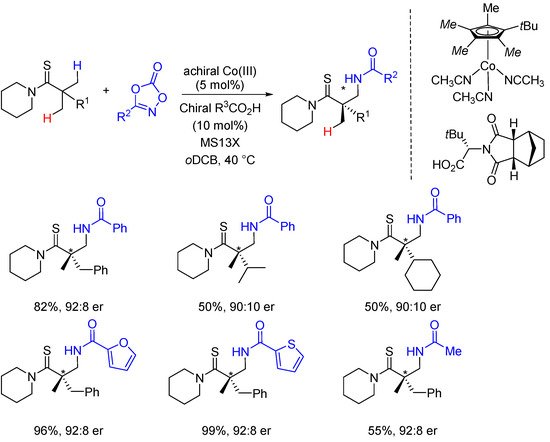
Scheme 6. Enantioselective thioamide C(sp3)-H amidations.
The enantio-discrimination step is an enantioselective concerted metalation deprotonation (CMD) step with the bulky chiral carboxylic acid (Scheme 7). Based on previously published DFT studies on an achiral version of the reaction [51][32] and mechanistic studies (H/D exchange), an irreversible C-H enantioselective deprotonation to give cyclo-cobalt complex B is advocated in these enantioselective C(sp3)-H amidations.
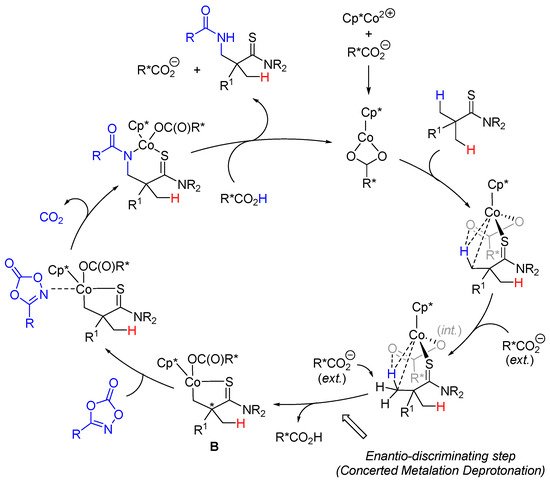
Scheme 7. Enantioselective thioamide C(sp3)-H amidations.
Quasi simultaneously [52][33], Chang described the Ir-catalyzed enantioselective intramolecular benzylic C-H amidation allowing the formation of γ-lactams in high yields and enantioselectivities [53,54][34][35]. The chiral diamine ligands employed in these transformations are commercially available, easily affording the Ir-based chiral catalysts. The reaction is very general being well suited with a broad range of substrates bearing prochiral Csp3-H bonds. High yields and very good selectivities were reached with substituted benzylic and aliphatic C-H bonds. Allylic and propargylic C-H bonds are also compatible albeit lower yields and selectivities are generally observed, alike when ortho substituted phenyl moieties were used (Scheme 8).
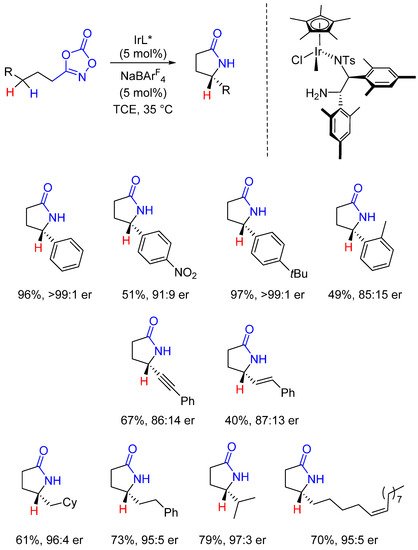
Scheme 8. Enantioselective prochiral C(sp3)-H amidations.
Beyond prochiral substrates, achiral substrates bearing a chirotopic carbon have also been used in desymmetrization intramolecular C-H amidations. Within this context, four compounds bearing two new and contiguous stereogenic centers were synthesized in high yields, dia- and enantioselectivities (two of them bearing a quaternary center) (Scheme 9).
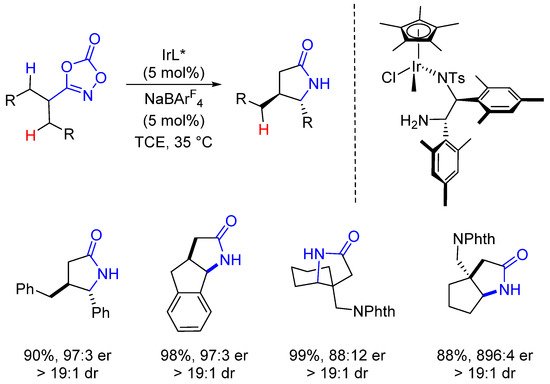
Scheme 9. Desymmetrization of enantiotopic substrates.
Mechanistically, two points deserve comments. The first one is about the need of N,N′-bidentate ligands to suppress the formation of isocyanate byproduct [55][36]. DFT calculations have also highlighted the crucial role of an intramolecular hydrogen bond in the enantiodiscrimination step (Scheme 10).
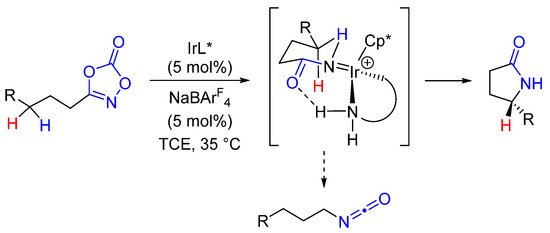
Scheme 10.
Mechanistic insights into Ir-catalyzed enantioselective formation of γ-lactams.
Chang, He and Chen next described the use of bulky N,N-bidentate aminoquinoline (AQ) α-amino-acid-based chiral ligands 2 leading to the δ-lactams at 20 °C in a HFIP/H2O mixture. (Scheme 11) [56][37]. With chiral ligand 2b, exceptional levels of selectivities have been observed with a wide range of substrates (benzylic, allylic, propargylic, alkyl, etc.). As illustrated in Scheme 11, the presence of the phthalimido-group (Phth) is crucial to ascertain high enantioselectivities. DFT studies have established that the Cp*, AQ and Phth groups are forming a pretty well defined hydrophobic chiral pocket fostering transition state organization in the aqueous polar solvent. Interestingly, the ligand 2b was obtained through a γ-C-H arylation from the corresponding tert-Leucine parent residue 2a.
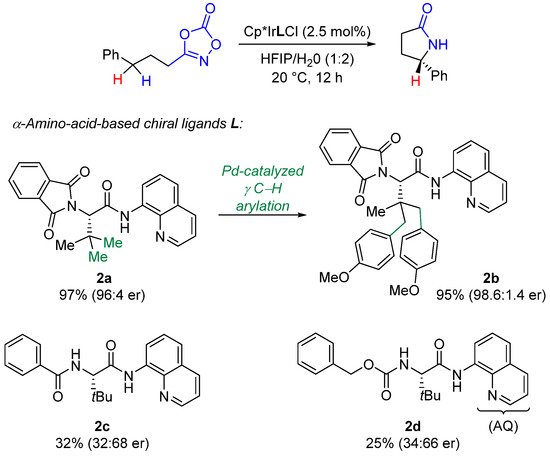
Scheme 11.
Chiral amino-acid-based ligands for enantioselective Ir-catalyzed C-H amidation.
Shortly after the publication of the seminal Chang’s paper, Yu and coworkers disclosed a related Ru-catalyzed intramolecular C-H amidation reaction in the presence of common chiral Noyori’s dpen (diphenylethylene diamine) ligands bearing electron-withdrawing aromatic groups [57][38]. As illustrated in Scheme 12, a collection of benzylic, allylic, propargylic and aliphatic C-H amidation products were obtained in moderate to high enantioselectivities.
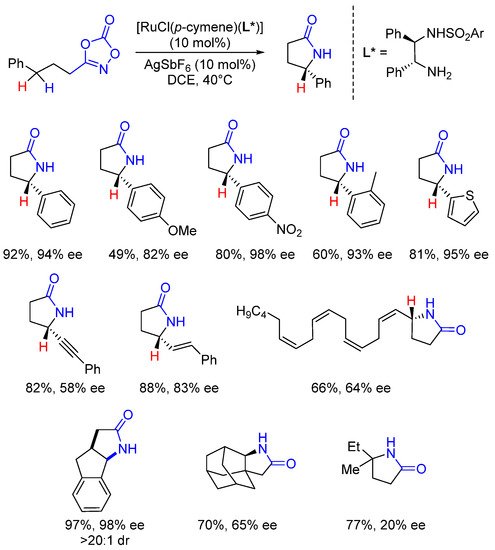
Scheme 12. Enantioselective Ru-catalyzed intramolecular amidations.
Finally, Meggers described the same type of Ru-catalyzed enantioselective intramolecular C-H amidations (Scheme 13). In this case, no chiral ligand or chiral carboxylic acid was employed to induce chirality but a chiral-at-metal ruthenium complex (for its structure, see Figure 2). Using non-C2-symmetric ruthenium catalyst I bearing remote NHC ligands, γ-lactams could be obtained in good yields and enantioselectivities at very low catalyst loading (0.1 mol%) [58,59][39][40].
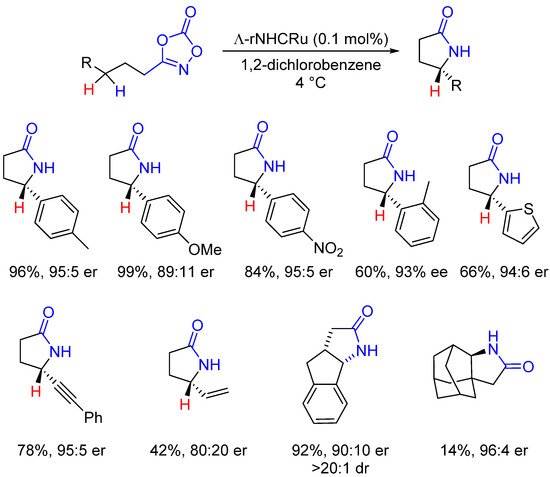
Scheme 13. Chiral-at-metal Ru catalysts for the synthesis of γ-lactams.
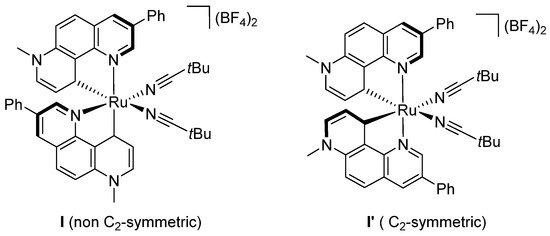
Figure 2.
Behavior of symmetric/non symmetric chiral-at-metal Ru catalysts in amidation reactions.
Interestingly, with the corresponding C2-symmetric diastereomer catalyst I’, a Curtius rearrangement occurs to give the corresponding undesired isocyanate as the major product (Figure 2). DFT studies conducted to understand such a striking different behavior have highlighted that both high strong electron-donating NHC ligand and the non-C2-symmetric structure of the catalyst account for the formation of an electron-rich nitrenoid-Ru intermediate, essential in the C-H amidation process.
Following their work on intermolecular C(sp3)-H enantioselective amidation of thioamides (vide supra), Matsunaga and Yoshino next described intermolecular C-H amidations via the differentiation of enantiotopic benzylic methylene C(sp3)-H bonds [60][41]. In this reaction, an achiral Cp*Rh(III) associated with a binaphtyl-based chiral carboxylic acid proved to be the best catalytic system to promote these intermolecular enantioselective C-H amidation reactions (Scheme 14). As previously observed and based on H/D exchange experiments, a carboxylate-assisted C-H activation is postulated to account for the observed enantioselectivities (see also Scheme 7).
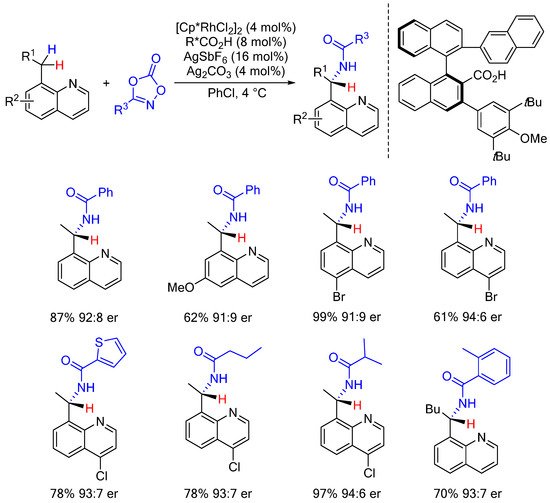
Scheme 14.
Enantioselective intermolecular C-H amidation of enantiotopic benzylic methylene.
Enantioselective C(sp3)-H amidations have been recently implemented by Blakey who described Rh-catalyzed regio- and enantioselective intermolecular allylic C-H amidation reactions (Scheme 15) [61][42]. The methodology proposed was based on the development and use of an original indenyl chiral rhodium ligand in charge of good regio- and enantioselectivities. Contrary to Cp/Cp* ligands that mainly acts through steric factors, the planar chiral indenyl ligand is believed to induce electronic asymmetry in the catalyst, playing on the ability of the indenyl ligand to open-up a different metal coordination by switching from η5- to η3-coordination [62][43]. The reaction displayed a quite broad scope concerning both dioxazolone and olefin substrates. DFT studies and the isolation of key intermediates have unveiled some key mechanistic details: (i) the reaction operates via the formation of a π-allyl complex and not through the direct C-H insertion of a Rh-nitrenoid species, (ii) the formation of the π-allyl rhodium complex is the rate- and enantio-determining step whereas (iii) the reductive C-N coupling from the nitrenoid rhodium intermediate appears to be the regio-determining step.
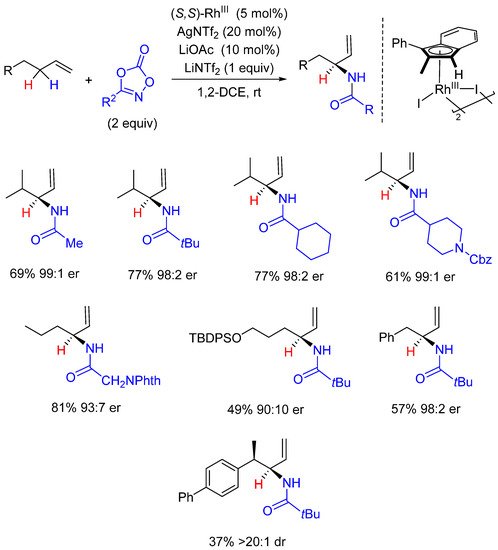
Scheme 15. Enantioselective allylic C-H amidation.
References
- Greenberg, A.; Breneman, C.M.; Liebman, J.F. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science; Wiley-Interscience: New York, NY, USA, 2000.
- Rajput, P.; Sharma, A. Synthesis and Biological Importance of Amide Analogues. J. Pharmacol. Med. Chem. 2018, 2, 22–31.
- de Figueiredo, R.M.; Suppo, J.-S.; Campagne, J.-M. Nonclassical Routes for Amide Bond Formation. Chem. Rev. 2016, 116, 12029–12122.
- Ojeda-Porras, A.; Gamba-Sánchez, D. Recent Developments in Amide Synthesis Using Nonactivated Starting Materials. J. Org. Chem. 2016, 81, 11548–11555.
- Park, Y.; Park, K.T.; Kim, J.G.; Chang, S. Mechanistic Studies on the Rh(III)-Mediated Amido Transfer Process Leading to Robust C-H Amination with a New Type of Amidating Reagent. J. Am. Chem. Soc. 2015, 137, 4534–4542.
- Park, Y.; Jee, S.; Kim, J.G.; Chang, S. Study of Sustainability and Scalability in the Cp*Rh(III)-Catalyzed Direct C-H Amidation with 1,4,2-Dioxazol-5-Ones. Org. Process Res. Dev. 2015, 19, 1024–1029.
- For a recent focus on dioxazolones, see: Van Vliet, K.M.; De Bruin, B. Dioxazolones: Stable Substrates for the Catalytic Transfer of Acyl Nitrenes. ACS Catal. 2020, 10, 4751–4769.
- For an early example of C-H amidation using stoechiometric [O]: Tsang, W.C.P.; Zheng, N.; Buchwald, S.L. Combined C-H Functionalization/C-N Bond Formation Route to Carbazoles. J. Am. Chem. Soc. 2005, 127, 14560–14561.
- For an early example of C-H amidation using stoechiometric [O]: Mei, T.S.; Wang, X.; Yu, J.Q. Pd(II)-Catalyzed Amination of C-H Bonds Using Single-Electron or Two-Electron Oxidants. J. Am. Chem. Soc. 2009, 131, 10806–10807.
- Park, J.; Chang, S. Comparative Catalytic Activity of Group 9 [CpMIII] Complexes: Cobalt-Catalyzed C-H Amidation of Arenes with Dioxazolones as Amidating Reagents. Angew. Chem. Int. Ed. 2015, 54, 14103–14107.
- Kweon, J.; Chang, S. Highly Robust Iron Catalyst System for Intramolecular C(sp3)−H Amidation Leading to γ-Lactams. Angew. Chem. Int. Ed. 2021, 60, 2909–2914.
- Park, S.H.; Kwak, J.; Shin, K.; Ryu, J.; Park, Y.; Chang, S. Amination Reaction Using Azides as the Nitrogen Source. J. Am. Chem. Soc. 2014, 136, 2492–2502.
- Carey, J.S.; Laffan, D.; Thomson, C.; Williams, M.T. Analysis of the Reactions Used for the Preparation of Drug Candidate Molecules. Org. Biomol. Chem. 2006, 4, 2337–2347.
- Roughley, S.D.; Jordan, A.M. The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem. 2011, 54, 3451–3479.
- Jeoung, D.; Kim, K.; Han, S.H.; Ghosh, P.; Lee, S.H.; Kim, S.; An, W.; Kim, H.S.; Mishra, N.K.; Kim, I.S. Phthalazinone-Assisted C–H Amidation Using Dioxazolones Under Rh(III) Catalysis. J. Org. Chem. 2020, 85, 7014–7023.
- Ghosh, P.; Samanta, S.; Hajra, A. Rhodium(III)-Catalyzed Ortho-C–H Amidation of 2-Arylindazoles with a Dioxazolone as an Amidating Reagent. Org. Biomol. Chem. 2020, 18, 1728–1732.
- Tang, S.-B.; Fu, X.-P.; Wu, G.-R.; Zhang, L.-L.; Deng, K.-Z.; Yang, J.-Y.; Xia, C.-C.; Ji, Y.-F. Rhodium(III)-Catalyzed C4-Amidation of Indole-Oximes with Dioxazolones via C–H Activation. Org. Biomol. Chem. 2020, 18, 7922–7931.
- Tian, X.; Li, X.; Duan, S.; Du, Y.; Liu, T.; Fang, Y.; Chen, W.; Zhang, H.; Li, M.; Yang, X. Room Temperature Benzofused Lactam Synthesis Enabled by Cobalt(III)-Catalyzed C(sp2)−H Amidation. Adv. Synth. Catal. 2021, 363, 1050–1058.
- Khake, S.M.; Chatani, N. The Direct Rh(III)-Catalyzed C–H Amidation of Aniline Derivatives Using a Pyrimidine Directing Group: The Selective Solvent Controlled Synthesis of 1,2-Diaminobenzenes and Benzimidazoles. Org. Lett. 2020, 22, 3655–3660.
- Dhiman, A.K.; Thakur, A.; Kumar, I.; Kumar, R.; Sharma, U. Co(III)-Catalyzed C–H Amidation of Nitrogen-Containing Heterocycles with Dioxazolones under Mild Conditions. J. Org. Chem. 2020, 85, 9244–9254.
- Zhang, J.; Xie, H.; Zhu, H.; Zhang, S.; Lonka, M.R.; Zou, H. Chameleon-like Behavior of the Directing Group in the Rh(III)-Catalyzed Regioselective C–H Amidation of Indole: An Experimental and Computational Study. ACS Catal. 2019, 9, 10233–10244.
- Wang, W.; Wu, J.; Kuniyil, R.; Kopp, A.; Nascimento Lima, R.; Ackermann, L. Peptide Late-Stage Diversifications by Rhodium-Catalyzed Tryptophan C7 Amidation. Chem 2020, 6, 3428–3439.
- Huang, J.; Ding, J.; Ding, T.-M.; Zhang, S.; Wang, Y.; Sha, F.; Zhang, F.-Y.; Wu, X.-Y.; Li, Q. Cobalt-Catalyzed Ortho-C(sp2)–H Amidation of Benzaldehydes with Dioxazolones Using Transient Directing Groups. Org. Lett. 2019, 21, 7342–7345.
- Kim, S.; Jeoung, D.; Kim, K.; Lee, S.B.; Lee, S.H.; Park, M.S.; Ghosh, P.; Mishra, N.K.; Hong, S.; Kim, I.S. Site-Selective C–H Amidation of 2-Aryl Quinazolinones Using Nitrene Surrogates. Eur. J. Org. Chem. 2020, 7134–7143.
- Scamp, R.J.; deRamon, E.; Paulson, E.K.; Miller, S.J.; Ellman, J.A. Cobalt(III)-Catalyzed C−H Amidation of Dehydroalanine for the Site-Selective Structural Diversification of Thiostrepton. Angew. Chem. Int. Ed. 2020, 59, 890–895.
- Lei, H.; Rovis, T. Ir-Catalyzed Intermolecular Branch-Selective Allylic C-H Amidation of Unactivated Terminal Olefins. J. Am. Chem. Soc. 2019, 141, 2268–2273.
- Knecht, T.; Mondal, S.; Ye, J.; Das, M.; Glorius, F. Intermolecular, Branch-Selective, and Redox-Neutral Cp*Ir III -Catalyzed Allylic C−H Amidation. Angew. Chem. Int. Ed. 2019, 58, 7117–7121.
- Burman, J.S.; Harris, R.J.; Farr, C.M.B.; Bacsa, J.; Blakey, S.B. Rh(III) and Ir(III)Cp∗ Complexes Provide Complementary Regioselectivity Profiles in Intermolecular Allylic C-H Amidation Reactions. ACS Catal. 2019, 9, 5474–5479.
- For a recent catalytic metal-free allylic C−H sulfonamidation strategy, see: Teh, W.P.; Obenschain, D.C.; Black, B.M.; Michael, F.E. Catalytic Metal-Free Allylic C−H Amination of Terpenoids. J. Am. Chem. Soc. 2020, 142, 16716–16722.
- Fukagawa, S.; Kato, Y.; Tanaka, R.; Kojima, M.; Yoshino, T.; Matsunaga, S. Enantioselective C(sp3)–H Amidation of Thioamides Catalyzed by a Cobalt III/Chiral Carboxylic Acid Hybrid System. Angew. Chem. Int. Ed. 2019, 58, 1153–1157.
- Sekine, D.; Ikeda, K.; Fukagawa, S.; Kojima, M.; Yoshino, T.; Matsunaga, S. Chiral 2-Aryl Ferrocene Carboxylic Acids for the Catalytic Asymmetric C(sp3)-H Activation of Thioamides. Organometallics 2019, 38, 3970–3978.
- For a racemic version of this reaction, see: Tan, P.W.; Mak, A.M.; Sullivan, M.B.; Dixon, D.J.; Seayad, J. Thioamide-Directed Cobalt(III)-Catalyzed Selective Amidation of C(sp3)-H Bonds. Angew. Chem. Int. Ed. 2017, 56, 16550–16554.
- Papers have been submitted in 24 October 2018 and in 5 November 2018 respectively.
- Park, Y.; Chang, S. Asymmetric Formation of γ-Lactams via C–H Amidation Enabled by Chiral Hydrogen-Bond-Donor Catalysts. Nat. Catal. 2019, 2, 219–227.
- Tan, T.D.; Ye, L.W. Chiral γ-lactam synthesis via asymmetric C–H amidation. Nat. Catal. 2019, 2, 182–183.
- Hong, S.Y.; Park, Y.; Hwang, Y.; Kim, Y.B.; Baik, M.H.; Chang, S. Selective Formation of γ-Lactams via C-H Amidation Enabled by Tailored Iridium Catalysts. Science 2018, 359, 1016–1021.
- Wang, H.; Park, Y.; Bai, Z.; Chang, S.; He, G.; Chen, G. Iridium-Catalyzed Enantioselective C(sp3)-H Amidation Controlled by Attractive Noncovalent Interactions. J. Am. Chem. Soc. 2019, 141, 7194–7201.
- Xing, Q.; Chan, C.M.; Yeung, Y.W.; Yu, W.Y. Ruthenium(II)-Catalyzed Enantioselective γ-Lactams Formation by Intramolecular C-H Amidation of 1,4,2-Dioxazol-5-Ones. J. Am. Chem. Soc. 2019, 141, 3849–3853.
- Zhou, Z.; Chen, S.; Hong, Y.; Winterling, E.; Tan, Y.; Hemming, M.; Harms, K.; Houk, K.N.; Meggers, E. Non- C2-Symmetric Chiral-at-Ruthenium Catalyst for Highly Efficient Enantioselective Intramolecular C(sp3)-H Amidation. J. Am. Chem. Soc. 2019, 141, 19048–19057.
- See also: Zhou, Z.; Tan, Y.; Yamahira, T.; Ivlev, S.; Xie, X.; Riedel, R.; Hemming, M.; Kimura, M.; Meggers, E. Enantioselective Ring-Closing C–H Amination of Urea Derivatives. Chem 2020, 6, 2024–2034.
- Fukagawa, S.; Kojima, M.; Yoshino, T.; Matsunaga, S. Catalytic Enantioselective Methylene C(sp3)-H Amidation of 8-Alkylquinolines Using a Cp*RhIII/Chiral Carboxylic Acid System. Angew. Chem. Int. Ed. 2019, 58, 18154–18158.
- Farr, C.M.B.; Kazerouni, A.M.; Park, B.; Poff, C.D.; Won, J.; Sharp, K.R.; Baik, M.H.; Blakey, S.B. Designing a Planar Chiral Rhodium Indenyl Catalyst for Regio- And Enantioselective Allylic C-H Amidation. J. Am. Chem. Soc. 2020, 142, 13996–14004.
- Obtained by chiral HPLC resolution of Rh(I)-COD mixture of enantiomers.