Quantifying O
6
-methylguanine-DNA methyltransferase (MGMT) promoter methylation plays an essential role in assessing the potential efficacy of alkylating agents in the chemotherapy of malignant gliomas. MGMT promoter methylation is considered to be a characteristic of subgroups of certain malignancies but has also been described in various peripheral inflammatory diseases. However, MGMT promoter methylation levels have not yet been investigated in non-neoplastic brain diseases. This study demonstrates for the first time that one can indeed detect slightly enhanced MGMT promoter methylation in individual cases of inflammatory demyelinating CNS diseases such as multiple sclerosis and progressive multifocal leucencephalopathy (PML), as well as in other demyelinating diseases such as central pontine and exptrapontine myelinolysis, and diseases with myelin damage such as Wallerian degeneration.
- DNA-methylation
- MGMT promoter methylation
- multiple sclerosis
- progressive multifocal leucencephalopathy (PML)
- central pontine and exptrapontine myelinolysis
- Wallerian degeneration
1. Introduction
O
6
-methylguanine-DNA methyltransferase (MGMT) is an important constitutively active enzyme which is expressed in every human cell, playing a pivotal role in the cellular defense against the toxicity of alkylating substances by removing methyl groups, particularly O
6-methylguanine residues, thereby repairing alkylated DNA and preventing mismatch errors during DNA replication [1]. Hypermethylation of the MGMT promoter region results in gene silencing, accompanied by decreased DNA repair, an effect that is seen in various tumors, including lung carcinoma, head and neck carcinomas, lymphoma, colorectal carcinoma, melanoma [1][2][3][4][5], as well as glioma [6] and particularly oligodendrogliomas [7]. MGMT hypermethylated gliomas are much more responsive to therapies with alkylating chemotherapeutics such as temozolomide (TMZ) than those without MGMT promoter hypermethylation [6][8]. MGMT promoter methylation seems to be at least partly the result of isocitrate dehydrogenase (IDH) mutations in gliomagenesis, since such gain-of-function mutations lead to the increased production of 2-hydroxyglutarate instead of alpha-ketoglutarate, which itself is an important co-factor for the proteins of the ten-eleven-translocation (TET) methylcytosine dioxygenases family such as TET1 and TET2 [9][10]. The TET family proteins are important DNA demethylases, and reduced levels of their co-factor alpha-ketoglutarate result in reduced enzyme activity, followed by globally increased DNA methylation in these cells, a phenomenon named G-CIMP [11].
-methylguanine residues, thereby repairing alkylated DNA and preventing mismatch errors during DNA replication [1]. Hypermethylation of the MGMT promoter region results in gene silencing, accompanied by decreased DNA repair, an effect that is seen in various tumors, including lung carcinoma, head and neck carcinomas, lymphoma, colorectal carcinoma, melanoma [1,2,3,4,5], as well as glioma [6] and particularly oligodendrogliomas [7]. MGMT hypermethylated gliomas are much more responsive to therapies with alkylating chemotherapeutics such as temozolomide (TMZ) than those without MGMT promoter hypermethylation [6,8]. MGMT promoter methylation seems to be at least partly the result of isocitrate dehydrogenase (IDH) mutations in gliomagenesis, since such gain-of-function mutations lead to the increased production of 2-hydroxyglutarate instead of alpha-ketoglutarate, which itself is an important co-factor for the proteins of the ten-eleven-translocation (TET) methylcytosine dioxygenases family such as TET1 and TET2 [9,10]. The TET family proteins are important DNA demethylases, and reduced levels of their co-factor alpha-ketoglutarate result in reduced enzyme activity, followed by globally increased DNA methylation in these cells, a phenomenon named G-CIMP [11].
In brain tissues with no evidence of any pathological alterations, the methylation rates of cytosine residues of the MGMT promoter region are described as not exceeding 3–4%, which in turn does not affect MGMT protein expression (so-called non-methylated MGMT promoter) [1][12]. Outside the central nervous system (CNS), MGMT promoter hypermethylation has been described in inflammatory diseases, especially in chronic inflammatory diseases of the gut, the liver or the colon [13][14]. In such cases, infections with oncogenic viruses, such as hepatitis-C-virus (HCV), Epstein–Barr virus (EBV) and hepatitis B virus (HBV) seem to be the critical event for MGMT promoter methylation [15][16][17][18]. Despite this knowledge, it has not yet been investigated whether MGMT promoter hypermethylation is present in non-neoplastic diseases of the CNS. The aim of our study is to investigate whether MGMT promoter methylation is a phenomenon that is restricted to neoplasms in the CNS, or whether it could be detected in other non-neoplastic CNS pathologies as well. Our study is, therefore, the first to analyze MGMT promoter methylation in a variety of non-neoplastic CNS diseases, and we report cases of variable MGMT hypermethylation in infectious, inflammatory and demyelinating CNS diseases, as well as in those resulting in damage to the myelin sheath.
In brain tissues with no evidence of any pathological alterations, the methylation rates of cytosine residues of the MGMT promoter region are described as not exceeding 3–4%, which in turn does not affect MGMT protein expression (so-called non-methylated MGMT promoter) [1,12]. Outside the central nervous system (CNS), MGMT promoter hypermethylation has been described in inflammatory diseases, especially in chronic inflammatory diseases of the gut, the liver or the colon [13,14]. In such cases, infections with oncogenic viruses, such as hepatitis-C-virus (HCV), Epstein–Barr virus (EBV) and hepatitis B virus (HBV) seem to be the critical event for MGMT promoter methylation [15,16,17,18]. Despite this knowledge, it has not yet been investigated whether MGMT promoter hypermethylation is present in non-neoplastic diseases of the CNS. The aim of our study is to investigate whether MGMT promoter methylation is a phenomenon that is restricted to neoplasms in the CNS, or whether it could be detected in other non-neoplastic CNS pathologies as well. Our study is, therefore, the first to analyze MGMT promoter methylation in a variety of non-neoplastic CNS diseases, and we report cases of variable MGMT hypermethylation in infectious, inflammatory and demyelinating CNS diseases, as well as in those resulting in damage to the myelin sheath.
2. The MGMT Promoter Is Variably Methylated in Various Non-Neoplastic CNS Diseases
We investigated autopsy and biopsy samples of healthy and diseased brains in terms of MGMT promoter methylation. Our analysis included brains without evidence of any pathological changes (“healthy controls”), as well as brains with infectious non-demyelinating diseases, i.e., bacterial, mycotic, inflammatory, viral (induced by HSV or HIV), or parasitic (toxoplasmosis), those with inflammatory-demyelinating diseases, i.e., multiple sclerosis (MS) and progressive multifocal leukoencephalopathy (PML), as well as those with non-neoplastic and non-inflammatory CNS conditions that damage the myelin sheaths, i.e., central pontine myelinolysis (CPM), extrapontine myelinolysis (EPM) and Wallerian degeneration. The degree of MGMT promoter methylation was compared with that in healthy controls (,Figure 1) as well as in hypermethylated gliomas. The mean percentage of methylated cytosine residues, measured at five CpG sites in each sample, was 5.6% in healthy controls (SD 0.88). The highest methylation rate in our controls was 6.6%. A sample was defined as “hypermethylated” if the mean methylation rate of all five CpG sites exceeded 7.7%, which was equivalent to the third quartile of the methylation rate in all non-neoplastic samples (
) as well as in hypermethylated gliomas (Supplementary Table S1). The mean percentage of methylated cytosine residues, measured at five CpG sites in each sample, was 5.6% in healthy controls (SD 0.88). The highest methylation rate in our controls was 6.6%. A sample was defined as “hypermethylated” if the mean methylation rate of all five CpG sites exceeded 7.7%, which was equivalent to the third quartile of the methylation rate in all non-neoplastic samples (, ).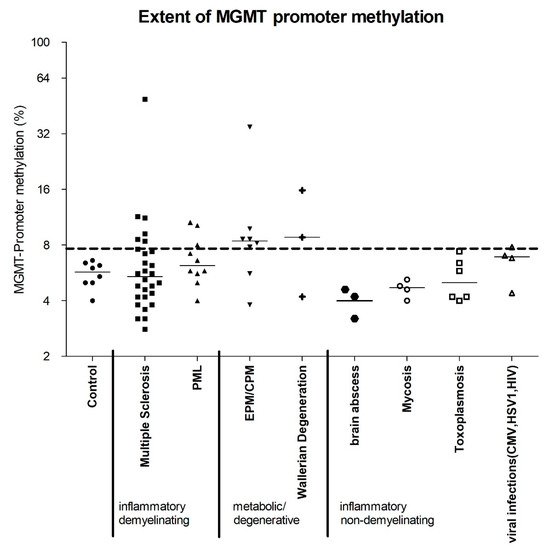
Figure 1.
n
n
n
n
n
n
n
n
n
n
n
n
n
n
n
Table 1.
6
| Case | MGMT Promoter Methylation (%) at Different Positions | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Age | Sex | Cause of Death | Disease Duration | Biopsy/Autopsy | Pos. 1 | Pos. 2 | Pos. 3 | Pos. 4 | Pos. 5 | Mean (1–5) | |
| Control | |||||||||||
| control 1 | 75 | f | multiorgan failure | na | autopsy | 2 | 6 | 5 | 7 | 5 | 5 |
| control 2 | 77 | f | sepsis | na | autopsy | 2 | 5 | 8 | 6 | 10 | 6.2 |
| control 3 | 61 | m | heart failure | na | autopsy | 4 | 5 | 6 | 7 | 10 | 6.4 |
| control 4 | 54 | f | respiratory failure | na | autopsy | 2 | 4 | 8 | 6 | 7 | 5.4 |
| control 5 | 57 | f | heart failure | na | autopsy | 3 | 4 | 9 | 7 | 10 | 6.6 |
| control 6 | 56 | m | multiorgan failure | na | autopsy | 2 | 5 | 7 | 4 | 12 | 6 |
| control 7 | 58 | m | heart failure | na | autopsy | 3 | 4 | 5 | 4 | 4 | 4 |
| control 8 | 78 | m | heart failure | na | autopsy | 4 | 5 | 6 | 5 | 5 | 5 |
| Multiple Sclerosis | |||||||||||
| MS autopsy 1 | 75 | f | pneumonia | autopsy | 5 | 5 | 9 | 7 | 16 | 8.4 | |
| MS autopsy 2 | 49 | m | pneumonia | autopsy | 2 | 4 | 4 | 2 | 4 | 3.2 | |
| MS autopsy 3 | 57 | f | respiratory failure | autopsy | 2 | 6 | 11 | 6 | 18 | 8.6 | |
| MS autopsy 4 | 52 | m | multiorgan failure | autopsy | 2 | 56 | 100 | 44 | 44 | 49.2 | |
| MS autopsy 5 | 68 | f | pneumonia | autopsy | 3 | 6 | 9 | 3 | 8 | 5.8 | |
| MS autopsy 6 | 62 | f | cachexia and pulmonary insufficiency | autopsy | 1 | 6 | 9 | 5 | 7 | 5.6 | |
| MS autopsy 7 | 44 | m | multiorgan failure | autopsy | 1 | 6 | 4 | 4 | 3 | 3.6 | |
| MS autopsy 8 | 57 | f | respiratory failure | autopsy | 9 | 15 | 10 | 8 | 14 | 11.2 | |
| MS autopsy 9 | 78 | f | stroke | autopsy | 9 | 10 | 12 | 12 | 14 | 11.4 | |
| MS autopsy 10 | 55 | m | respiratory insufficiency complicating pneumonia and urosepsis | autopsy | 4 | 6 | 7 | 5 | 10 | 6.4 | |
| MS autopsy 11 | 56 | f | respiratory insufficiency in pneumonia | autopsy | 3 | 6 | 4 | 6 | 7 | 5.2 | |
| MS autopsy 12 | 44 | m | aspiration pneumonia | autopsy | 2 | 4 | 9 | 7 | 9 | 6.2 | |
| MS autopsy 13 | 63 | m | pneumonia | autopsy | 6 | 9 | 5 | 6 | 10 | 7.2 | |
| MS autopsy 14 | 53 | m | assisted suicide | autopsy | 4 | 3 | 4 | 3 | 5 | 3.8 | |
| MS autopsy 15 | 54 | f | heart failure | autopsy | 3 | 4 | 5 | 2 | 7 | 4.2 | |
| MS autopsy 16 | 48 | f | respiratory failure | autopsy | 4 | 6 | 9 | 8 | 11 | 7.6 | |
| MS autopsy 17 | 58 | m | terminal renal failure | autopsy | 4 | 7 | 3 | 3 | 11 | 5.6 | |
| MS autopsy 18 | 66 | f | cancer metastases in the liver resulting in severe failure of the liver functions | autopsy | 3 | 4 | 5 | 3 | 8 | 4.6 | |
| MS autopsy 19 | 56 | f | respiratory insufficiency in pneumonia | autopsy | 6 | 7 | 9 | 7 | 8 | 7.4 | |
| MS autopsy 20 | 26 | f | multiorgan failure | autopsy | 6 | 8 | 9 | 8 | 15 | 9.2 | |
| MS biopsy 1 | 43 | m | na | na | biopsy | 3 | 4 | 5 | 3 | 6 | 4.2 |
| MS biopsy 2 | 35 | f | na | 1 month | biopsy | 3 | 5 | 4 | 2 | 5 | 3.8 |
| MS biopsy 3 | 9 | m | na | <1 month | biopsy | 4 | 5 | 4 | 4 | 8 | 5 |
| MS biopsy 4 | 25 | f | na | na | biopsy | 3 | 4 | 6 | 4 | 7 | 4.8 |
| MS biopsy 5 | 31 | m | na | na | biopsy | 2 | 4 | 6 | 4 | 8 | 4.8 |
| MS biopsy 6 | 46 | f | na | <1 month | biopsy | 2 | 3 | 3 | 3 | 3 | 2.8 |
| MS biopsy 7 | 61 | m | na | na | biopsy | 4 | 5 | 5 | 4 | 4 | 4.4 |
| MS biopsy 8 | 35 | f | na | 15 years | biopsy | 3 | 3 | 4 | 3 | 3 | 3.2 |
| PML | |||||||||||
| PML 1 | 34 | m | na | na | biopsy | 4 | 6 | 8 | 4 | 6 | 5.6 |
| PML 2 | 31 | m | na | na | biopsy | 6 | 6 | 7 | 6 | 8 | 6.6 |
| PML 3 | 41 | m | renal failure | na | autopsy | 6 | 8 | 11 | 8 | 7 | 8 |
| PML 4 | 51 | m | na | na | autopsy | 8 | 13 | 7 | 12 | 13 | 10.6 |
| PML 5 | 65 | f | na | na | autopsy | 7 | 8 | 15 | 10 | 11 | 10.2 |
| PML 6 | 58 | f | na | na | autopsy | 3 | 6 | 6 | 3 | 7 | 5 |
| PML 7 | 77 | f | na | na | biopsy | 4 | 6 | 7 | 5 | 7 | 5.8 |
| PML 8 | 66 | f | na | na | biopsy | 3 | 5 | 7 | 5 | 9 | 5.8 |
| PML 9 | 41 | m | renal failure | na | autopsy | 4 | 6 | 8 | 6 | 12 | 7.2 |
| PML 10 | 59 | f | na | na | biopsy | 3 | 4 | 5 | 4 | 4 | 4 |
| CPM/EPM | |||||||||||
| CPM 1 | 54 | m | sepsis | na | autopsy | 6 | 6 | 10 | 6 | 15 | 8.6 |
| CPM 2 | 52 | f | na | na | autopsy | 5 | 8 | 6 | 7 | 17 | 8.6 |
| CPM 3 | 54 | m | sepsis | na | autopsy | 5 | 7 | 10 | 7 | 12 | 8.2 |
| CPM 4 | 52 | f | na | na | autopsy | 23 | 35 | 30 | 44 | 42 | 34.8 |
| CPM 5 | 53 | m | CPM, cerebral hemorrhage | na | autopsy | 3 | 5 | 3 | 4 | 4 | 3.8 |
| CPM 6 | 86 | m | CPM | na | autopsy | 7 | 6 | 7 | 5 | 3 | 5.6 |
| CPM 7 | 41 | m | CPM, cerebral hemorrhage | na | autopsy | 3 | 7 | 10 | 9 | 10 | 7.8 |
| CPM 8 | 55 | m | stroke | na | autopsy | 8 | 10 | 11 | 9 | 11 | 9.8 |
| Wallerian degeneration | |||||||||||
| WAL 1 | 59 | m | multiorgan failure | na | autopsy | 12 | 19 | 13 | 14 | 21 | 15.8 |
| WAL 2 | 50 | m | central regulatory failure | na | autopsy | 5 | 7 | 12 | 7 | 13 | 8.8 |
| WAL 3 | 65 | m | stroke | na | autopsy | 2 | 5 | 5 | 2 | 7 | 4.2 |
| brain abscess | |||||||||||
| ABS 1 | 42 | m | na | na | biopsy | 2 | 4 | 4 | 2 | 4 | 3.2 |
| ABS 2 | 45 | m | na | na | biopsy | 3 | 4 | 5 | 4 | 5 | 4.2 |
| ABS 3 | 3 | f | na | na | biopsy | 3 | 5 | 5 | 4 | 6 | 4.6 |
| mycosis | |||||||||||
| Myc 1 | 55 | m | na | na | autopsy | 2 | 4 | 5 | 4 | 5 | 4 |
| Myc 2 | 48 | f | na | na | biopsy | 3 | 5 | 5 | 4 | 6 | 4.6 |
| Myc 3 | 56 | f | stroke, sepsis | na | autopsy | 4 | 4 | 7 | 4 | 7 | 5.2 |
| Myc 4 | 84 | f | na | na | biopsy | 4 | 5 | 5 | 4 | 6 | 4.8 |
| toxoplasmosis | 6 | 6 | 9 | 7 | 9 | 7.4 | |||||
| Toxo 1 | 59 | f | na | na | biopsy | 3 | 3 | 6 | 4 | 5 | 4.2 |
| Toxo 2 | 47 | m | na | na | biopsy | 6 | 5 | 8 | 7 | 6 | 6.4 |
| Toxo 3 | 64 | m | na | na | biopsy | 3 | 3 | 5 | 4 | 6 | 4.2 |
| Toxo 4 | 29 | f | na | na | biopsy | 3 | 4 | 4 | 4 | 5 | 4 |
| Toxo 5 | 40 | m | na | na | biopsy | 5 | 6 | 8 | 3 | 7 | 5.8 |
| Toxo 6 | 22 | m | na | na | biopsy | 6 | 6 | 9 | 7 | 9 | 7.4 |
| CMV | |||||||||||
| CMV | 38 | m | na | autopsy | 6 | 5 | 12 | 3 | 8 | 6.8 | |
| HSV | |||||||||||
| HSV | 37 | m | HSV-encephalitis | 4 weeks | autopsy | 1 | 6 | 4 | 6 | 5 | 4.4 |
| HIV | |||||||||||
| HIV 1 | 44 | m | respiratory failure | na | autopsy | 4 | 7 | 9 | 5 | 10 | 7 |
| HIV 2 | 51 | m | multiorgan failure | na | autopsy | 5 | 8 | 8 | 7 | 11 | 7.8 |
Abbreviations: m, male; f, female; na, not applicable; PML: progressive multifocal leucencephalopathy; CPM: central pontine myelinolysis; EPM: extrapontine myelinolysis.
p
= 0.82; sex: Pearsons r = 0.15;p
= 0.18) or with the total number of apoptotic cells measured by immunohistochemical staining for caspase-3 (Pearsons r = 0.23,p
= 0.24). In order to rule out any influence of cause of death on the extent of methylation, we categorized our autopsy samples into five groups concerning their cause of death, namely (1) multiorgan failure (n
= 7), (2) sepsis (n
= 4), (3) cardiac reasons (n
= 5), (4) pulmonary reasons (n
= 14) and (5) others (n
= 13). The means of methylation of those groups were compared to each other via ANOVA afterwards. We did not find any significant differences between the groups (p
> 0.05). Therefore, the (reason of) death itself does not seem to affect MGMT methylation. It is worth mentioning that MGMT promoter methylation rates differed significantly between samples obtained from MS patients by biopsy or autopsy (biopsy: mean 4.13%, SD 0.8 vs. autopsy: mean 8.72%, SD 9.8;p
< 0.01). All deceased MS patients whose autopsy samples we analyzed had suffered from long-standing chronic MS, whereas most patients who had been diagnosed with MS by brain biopsy had a short history of symptoms and disease course (i.e., a few months maximum; the only exception was MS sample 8). In each case, the biopsy had been performed because a tumor had been suspected. Therefore, it seems plausible that MGMT promoter methylation is associated with long-standing and chronic MS disease rather than with more acute and active forms. Of note, the autopsy samples were from MS patients significantly older than those who had had a brain biopsy (autopsy: mean 56.1 years, SD 11.5 vs. biopsy: mean 35.6 years, SD 15.4;p
< 0.01). However, since there was no association between age and MGMT promoter methylation across all analyzed samples it is much more likely that the activity status of the disease is responsible for the differences in MGMT methylation. Since 50% of the hypermethylated samples (ten out of 20 samples) were linked to non-inflammatory diseases, such as CPM, EPM and Wallerian degeneration, an association of MGMT promoter methylation to inflammatory infiltrate per se and, in particular, to specific inflammatory cells (e.g., lymphocytes, granulocytes, plasma cells) was not expected. However, since injured axonal networks display the fundamental commonality of all hypermethylated samples, we suspected a link between axonal damage and MGMT promoter methylation. SMI31 is a marker for phosphorylated neurofilaments which display the integrity of axonal networks [19]. We therefore stained all samples against SMI31 and correlated the density of SMI31-positive phosphorylated neurofilaments to MGMT promoter methylation rate. Nonetheless, we were unable to verify our hypothesis in this investigation (Pearsons r = 0.05;p
= 0.78). However, this negative result might be due to relatively small differences in methylation levels compared with healthy controls, as well as a heterogeneity of hypermethylation and non-methylation within one disease entity. Since the degree of hypermethylation was relatively low, we next wanted to investigate whether the slight differences might be reflected in lowered MGMT mRNA and protein expression levels. To begin with, we attempted to quantify MGMT mRNA levels in formalin fixed paraffin embedded (FFPE) samples from healthy controls and those with hypermethylation using qPCR. Suitable results were only obtained for a few samples (healthy control (control 1–4); CPM/EPM: CPM 2, Wallerian degeneration: WAL 2, WAL 3), and a reliable statistical assessment was not possible due to the small sample size. Nonetheless, MGMT mRNA levels tended to be lower in hypermethylated samples compared with healthy controls (Wallerian degeneration: 1.36-fold lower levels, CPM/EPM: four-fold lower levels) . In a next step, all samples were stained immunohistochemically against MGMT for further analysis of MGMT protein expression levels. For the quantification of MGMT expression in glial cells, we automatically measured the numbers of MGMT-positive glial cells per mm2
. Of note, small differences in methylation rates of the MGMT promoter resulted in significantly lower MGMT protein expression levels (). Healthy controls (C) showed the constitutive expression of MGMT in almost all glial cells, i.e., astrocytes and particularly in oligodendrocytes, as well as in neurons, while the staining intensity of glial cells was significantly reduced in all samples with hypermethylated MGMT promoter (D–H) (p
< 0.01, Wallerian degeneration not significant due to small sample size). Indeed, the hypermethylated samples showed only scattered MGMT-expressing glial cells in the areas of the lesions (D–H), while the neurons showed stable staining intensities across all diagnostic groups (C–H, insets), serving as an internal positive control and thereby ruling out staining artifacts. The number of MGMT-positive glial cells was found to be independent of the extent of myelination in MS lesions (chronic inactive plaque (CIAP) vs. remyelinated shadow plaques) (D,E) (no significant difference between the groups).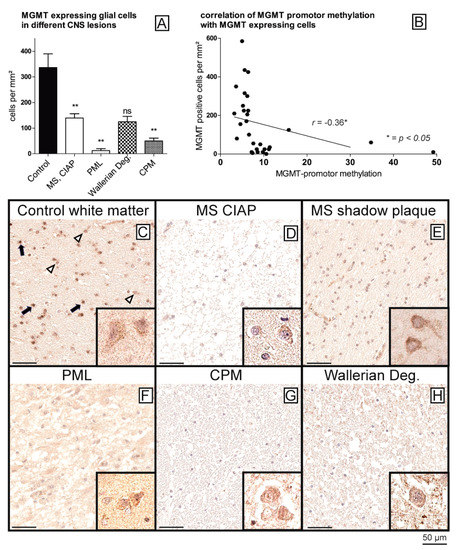
Figure 2.
p
A
B
p
C
D
E
F
G
H
D
E
C
H
3. Discussion
MGMT is a protein that plays an important role in DNA repair and cellular defense against toxic agents such as alkylating substances. It catalyzes the transfer of methyl groups from mainly O
6
-methylguanine but also O
4-methylguanine to cysteine residues of its own molecule, thereby repairing damaged DNA [1]. MGMT promoter methylation is a common phenomenon in inflammatory diseases outside the CNS and is often associated with oncogenic viruses (e.g., HBV, EBV, HCV) [15][16][17][18]. Moreover, MGMT promoter hypermethylation had been described in precancerous lesions such as colitis ulcerosa and Crohn’s disease but also in gastritis [13][20] as well as in various cancers, including colorectal carcinoma, lung carcinoma, lymphoma, melanoma, and, in the CNS in glioma, particularly oligodendroglioma but also in IDH-mutated as well as in IDH-wildtype astrocytoma [5][6]. Previously, it was postulated that DNA hypermethylation is a phenomenon attributed to cancers or to malignant transformation of precancerous lesions and that consequently it could be used as a tumor biomarker [21]. Up to now, only a few studies have focused on MGMT promoter methylation in the CNS beyond cancerous brain tissues. Hsu et al. investigated healthy brains and brain biopsies obtained during epilepsy surgery and found that none of their samples exhibited MGMT promoter hypermethylation [12]. A single study analyzed the promoter methylation rates of different DNA repairing enzymes, including MGMT, in Alzheimer’s disease and also did not find the MGMT promoter to be hypermethylated [22]. Thus, both studies underscored the hypothesis [21] that MGMT methylation can be attributed to CNS neoplasms rather than to non-neoplastic CNS diseases. Nonetheless, other CNS diseases, particularly of infectious and inflammatory nature have not yet been studied. To our knowledge, we were the first to show that MGMT promoter hypermethylation takes place in a variety of non-neoplastic CNS diseases. While we were not able to attribute MGMT methylation to single disease entities or distinct pathogens, it is noteworthy that we did not find any associations with infectious non-demyelinating diseases (i.e., bacterial or mycotic abscesses, toxoplasmosis, viral encephalitis), a fact that is in contrast to the findings outside the CNS. However, we found MGMT promoter hypermethylation in the samples from patients with diseases associated with disintegrated myelin sheaths, i.e., inflammatory and demyelinating (MS and PML), as well as non-inflammatory metabolic or degenerative CNS diseases (CPM/EPM/Wallerian degeneration). These results, of course, reduce the hypothesized specificity of MGMT promoter methylation for neoplastic processes [21].
-methylguanine to cysteine residues of its own molecule, thereby repairing damaged DNA [1]. MGMT promoter methylation is a common phenomenon in inflammatory diseases outside the CNS and is often associated with oncogenic viruses (e.g., HBV, EBV, HCV) [15,16,17,18]. Moreover, MGMT promoter hypermethylation had been described in precancerous lesions such as colitis ulcerosa and Crohn’s disease but also in gastritis [13,26] as well as in various cancers, including colorectal carcinoma, lung carcinoma, lymphoma, melanoma, and, in the CNS in glioma, particularly oligodendroglioma but also in IDH-mutated as well as in IDH-wildtype astrocytoma [5,6]. Previously, it was postulated that DNA hypermethylation is a phenomenon attributed to cancers or to malignant transformation of precancerous lesions and that consequently it could be used as a tumor biomarker [27]. Up to now, only a few studies have focused on MGMT promoter methylation in the CNS beyond cancerous brain tissues. Hsu et al. investigated healthy brains and brain biopsies obtained during epilepsy surgery and found that none of their samples exhibited MGMT promoter hypermethylation [12]. A single study analyzed the promoter methylation rates of different DNA repairing enzymes, including MGMT, in Alzheimer’s disease and also did not find the MGMT promoter to be hypermethylated [28]. Thus, both studies underscored the hypothesis [27] that MGMT methylation can be attributed to CNS neoplasms rather than to non-neoplastic CNS diseases. Nonetheless, other CNS diseases, particularly of infectious and inflammatory nature have not yet been studied. To our knowledge, we were the first to show that MGMT promoter hypermethylation takes place in a variety of non-neoplastic CNS diseases. While we were not able to attribute MGMT methylation to single disease entities or distinct pathogens, it is noteworthy that we did not find any associations with infectious non-demyelinating diseases (i.e., bacterial or mycotic abscesses, toxoplasmosis, viral encephalitis), a fact that is in contrast to the findings outside the CNS. However, we found MGMT promoter hypermethylation in the samples from patients with diseases associated with disintegrated myelin sheaths, i.e., inflammatory and demyelinating (MS and PML), as well as non-inflammatory metabolic or degenerative CNS diseases (CPM/EPM/Wallerian degeneration). These results, of course, reduce the hypothesized specificity of MGMT promoter methylation for neoplastic processes [27].
Our results show that the spectrum of MGMT promoter methylation in healthy controls is relatively narrow with a range of 4 to 6.6 percent, which is in accordance with those of Christmann et al. and Hsu and colleagues [1][12]. They underscore the finding that healthy brain tissue is apparently never hypermethylated and that MGMT promoter methylation is only associated with severe brain diseases. The fact that MGMT hypermethylation was especially found in autopsy samples from patients with long-standing, chronic MS, rather than in biopsies where the disease was more active in nature, leads to the hypothesis that MGMT promoter methylation might be associated with later disease stages accompanied by axonal damage. This hypothesis is underscored by various studies that linked MGMT methylation to later disease stages. For example, Alvarez et al. demonstrated MGMT hypermethylation in chronic, but not in early stages of
Our results show that the spectrum of MGMT promoter methylation in healthy controls is relatively narrow with a range of 4 to 6.6 percent, which is in accordance with those of Christmann et al. and Hsu and colleagues [1,12]. They underscore the finding that healthy brain tissue is apparently never hypermethylated and that MGMT promoter methylation is only associated with severe brain diseases. The fact that MGMT hypermethylation was especially found in autopsy samples from patients with long-standing, chronic MS, rather than in biopsies where the disease was more active in nature, leads to the hypothesis that MGMT promoter methylation might be associated with later disease stages accompanied by axonal damage. This hypothesis is underscored by various studies that linked MGMT methylation to later disease stages. For example, Alvarez et al. demonstrated MGMT hypermethylation in chronic, but not in early stages of
Helicobacter pylori-associated gastritis [23], and the study of N. Zekri et al. highlighted that long-standing chronic Hepatits C infection with progression to hepatocellular carcinoma is accompanied by promoter methylation of APC gene in early, and that of MGMT in later disease stages [24]. Furthermore, MGMT methylation is seen in COPD and lung cancers due to chronic tobacco abuse [25].
-associated gastritis [29], and the study of N. Zekri et al. highlighted that long-standing chronic Hepatits C infection with progression to hepatocellular carcinoma is accompanied by promoter methylation of APC gene in early, and that of MGMT in later disease stages [30]. Furthermore, MGMT methylation is seen in COPD and lung cancers due to chronic tobacco abuse [31].
This hypothesis of MGMT methylation in later disease stages could explain the great heterogeneity in terms of MGMT methylation levels in the individual samples from our cohort. This mixture of hypermethylation and non-methylation within one disease entity (as observed in our samples) is a well-known phenomenon, which has been described in chronic hepatitis C infected and fibrotic livers [15], Hodgkin lymphomas [17], brain metastases of various cancers (reviewed in [1]), glioblastoma [26] as well as in our glioma samples. This underscores that MGMT promoter methylation shows a wide range of methylation within one disease entity, while the exact cause of this broad spectrum remains unknown. Nonetheless, we are aware that our total numbers of samples from the various disease entities were small and that our results might thus not be generalizable. As a consequence of our relatively small sample sizes, MGMT promoter methylation rates did—of course—not reach statistical significance, since this would only be possible if we could have investigated hundreds of samples. Bearing in mind that those disease entities investigated here are relatively rare and biopsy or autopsy material is even rarer, we really believe that—although not reaching statistical significance—our collection is a relatively big one and that our results are anyhow of great interest.
This hypothesis of MGMT methylation in later disease stages could explain the great heterogeneity in terms of MGMT methylation levels in the individual samples from our cohort. This mixture of hypermethylation and non-methylation within one disease entity (as observed in our samples) is a well-known phenomenon, which has been described in chronic hepatitis C infected and fibrotic livers [15], Hodgkin lymphomas [17], brain metastases of various cancers (reviewed in [1]), glioblastoma [32] as well as in our glioma samples. This underscores that MGMT promoter methylation shows a wide range of methylation within one disease entity, while the exact cause of this broad spectrum remains unknown. Nonetheless, we are aware that our total numbers of samples from the various disease entities were small and that our results might thus not be generalizable. As a consequence of our relatively small sample sizes, MGMT promoter methylation rates did—of course—not reach statistical significance, since this would only be possible if we could have investigated hundreds of samples. Bearing in mind that those disease entities investigated here are relatively rare and biopsy or autopsy material is even rarer, we really believe that—although not reaching statistical significance—our collection is a relatively big one and that our results are anyhow of great interest.
The differences in methylation between the hypermethylated samples and those of healthy controls were often relatively small. This might have been due to the fact that we used whole slides for our analysis and did not perform any macrodissection of the lesions or microdissection of individual cells. While we believed that analyzing the lesion as well as the surrounding brain microenvironment would give more information than assessing the lesion or individual cells alone, this approach may have diminished the magnitude of our detected effects. Nonetheless, we were able to show reduced MGMT mRNA and especially protein expression levels in hypermethylated samples despite the low-level methylation differences. This is a well-known effect since small differences in DNA methylation are accompanied by altered protein expression levels, as has been shown in rheumatoid arthritis and in the frontal cortices of Alzheimer’s disease patients [27][28].
The differences in methylation between the hypermethylated samples and those of healthy controls were often relatively small. This might have been due to the fact that we used whole slides for our analysis and did not perform any macrodissection of the lesions or microdissection of individual cells. While we believed that analyzing the lesion as well as the surrounding brain microenvironment would give more information than assessing the lesion or individual cells alone, this approach may have diminished the magnitude of our detected effects. Nonetheless, we were able to show reduced MGMT mRNA and especially protein expression levels in hypermethylated samples despite the low-level methylation differences. This is a well-known effect since small differences in DNA methylation are accompanied by altered protein expression levels, as has been shown in rheumatoid arthritis and in the frontal cortices of Alzheimer’s disease patients [33,34].
Chronic gastritis induced by
Helicobacter pylori showed MGMT promoter hypermethylation based on accelerated NF-κB signaling [29]. The pro-inflammatory NF-κB pathway plays a pivotal role in various inflammatory diseases [30]. In MS, NF-κB is essentially involved not only in the activation of peripheral immune cells but also in reactive processes of brain-derived cells [31]. Upregulated NF-κB activity has also been detected in PML and Wallerian degeneration [32][33].
showed MGMT promoter hypermethylation based on accelerated NF-κB signaling [35]. The pro-inflammatory NF-κB pathway plays a pivotal role in various inflammatory diseases [36]. In MS, NF-κB is essentially involved not only in the activation of peripheral immune cells but also in reactive processes of brain-derived cells [37]. Upregulated NF-κB activity has also been detected in PML and Wallerian degeneration [38,39].
A fundamental similarity of all hypermethylated samples is the damage to the myelin sheath. Oligodendrocytes are glial cells that are responsible for producing myelin and for effective remyelination. Different DNA methylation patterns are involved in oligodendrocyte differentiation, myelin production and remyelination after injury. High TET1 expression levels seem to be required for efficient remyelination after myelin damage [34]. A recent study reported that demyelinated lesions in the hippocampus of MS patients showed upregulated levels of DNA methyl transferases (DNMTs)—enzymes that are responsible for DNA methylation—while TET levels were downregulated [35]. We have also found reduced TET1 protein expression levels in all our samples with MGMT promoter hypermethylation, leading to the hypothesis that there is an imbalance between DNA methylation and DNA demethylation at least by reduced DNA demethylation. Whether DNA methylation is additionally enhanced by upregulated DNMT expression levels as already shown for MS [35] has to be investigated in future studies. Against this background, it is possible that the MGMT promoter, as well as general DNA methylation in other DNA regions, displays an essential epigenetic mechanism for the defective myelin regeneration in CNS diseases with myelin damage and loss. Indeed, it was shown that promoters of genes such as BCL2L2 and NDRG1, both genes that regulate oligodendrocyte survival, are hypermethylated in normal appearing white matter of MS patients, and that consequently the proteins are less expressed, which in turn leads to accelerated oligodendrocyte apoptosis, accompanied by less efficient remyelination [36].
A fundamental similarity of all hypermethylated samples is the damage to the myelin sheath. Oligodendrocytes are glial cells that are responsible for producing myelin and for effective remyelination. Different DNA methylation patterns are involved in oligodendrocyte differentiation, myelin production and remyelination after injury. High TET1 expression levels seem to be required for efficient remyelination after myelin damage [20]. A recent study reported that demyelinated lesions in the hippocampus of MS patients showed upregulated levels of DNA methyl transferases (DNMTs)—enzymes that are responsible for DNA methylation—while TET levels were downregulated [40]. We have also found reduced TET1 protein expression levels in all our samples with MGMT promoter hypermethylation, leading to the hypothesis that there is an imbalance between DNA methylation and DNA demethylation at least by reduced DNA demethylation. Whether DNA methylation is additionally enhanced by upregulated DNMT expression levels as already shown for MS [40] has to be investigated in future studies. Against this background, it is possible that the MGMT promoter, as well as general DNA methylation in other DNA regions, displays an essential epigenetic mechanism for the defective myelin regeneration in CNS diseases with myelin damage and loss. Indeed, it was shown that promoters of genes such as BCL2L2 and NDRG1, both genes that regulate oligodendrocyte survival, are hypermethylated in normal appearing white matter of MS patients, and that consequently the proteins are less expressed, which in turn leads to accelerated oligodendrocyte apoptosis, accompanied by less efficient remyelination [41].
Further and still larger scale studies with whole genome methylation analyses should be performed to better understand the influence of DNA methylation on de- and remyelination processes in MS and other demyelinating diseases. It is known that cell type-specific methylation patterns exist [34]. It would, therefore, be important to also investigate the relation of oligodendrocytes or astrocytes to possible DNA hypermethylation in further studies.
Further and still larger scale studies with whole genome methylation analyses should be performed to better understand the influence of DNA methylation on de- and remyelination processes in MS and other demyelinating diseases. It is known that cell type-specific methylation patterns exist [20]. It would, therefore, be important to also investigate the relation of oligodendrocytes or astrocytes to possible DNA hypermethylation in further studies.
In summary, we have shown for the first time that MGMT hypermethylation is indeed found in non-neoplastic CNS diseases showing damage of the myelin sheath due to various conditions. Thus, we demonstrated that MGMT methylation is not restricted to neoplasms or strictly associated to distinct pathogens, as well as oncogenic viruses or bacteria. Therefore, the capability of MGMT hypermethylation to serve as a biomarker for neoplasms or as indicator of malignant transformation of precancerous lesions is highly reduced by our results. The fact that MGMT methylation is found in chronic processes that lead to myelin loss and consequently to axonal damage, shed light into possible epigenetic and pathophysiological processes involved in demyelination and might thus offer new therapeutic opportunities in the future.