Cancer-associated fibroblasts (CAFs) are prominent and key components of the TME in most types of solid tumors. Extensive research over the past decade revealed their ability to modulate cancer metastasis, angiogenesis, tumor mechanics, immunosuppression, and drug access through synthesis and remodeling of the extracellular matrix and production of growth factors. Thus, they are considered to impede the response to current clinical cancer therapies. Therefore, targeting CAFs to counteract these protumorigenic effects, and overcome the resistance to current therapeutic options, is an appealing and emerging strategy.
- cancer associated fibroblasts
- therapy resistance
- lung cancer
- pancreatic cancer
1. Introduction
As our knowledge on cancer biology continuously evolves, the tumor microenvironment (TME) has gained interest over the past decade. Before, cancer research has been focusing primarily on the malignant cell itself [1]. However, cancers are not simply autonomous neoplastic cells [1] but interact with a complex surrounding ecosystem of stromal cells, known as the TME [2]. The TME consists of cancer-associated fibroblasts (CAFs), extracellular matrix (ECM), endothelial cells, and infiltrating innate and adaptive immune cells (e.g., natural killer (NK) cells and T cells, respectively) [3]. CAFs, the most prominent [4] and key component [5] of the TME, can either originate from normal fibroblasts, which are non-epithelial, non-endothelial, non-immune resting mesenchymal cells in diverse connective tissue components [6], or other precursor cells such as bone marrow-derived mesenchymal stem cells, epithelial cells, carcinoma cells, endothelial cells, pericytes, smooth muscle cells, adipocytes, fibrocytes, or specialized cells such as pancreatic stellate cells (PSCs) [3]. However, as there are no unique markers for fibroblasts that are not expressed by other cell types, it is hard to define a CAF [3][7]. Therefore, all fibroblasts associated with tumors [6] that are negative for epithelial, endothelial, and leukocyte markers with an elongated morphology and lacking the mutations found within cancer cells might be considered CAFs [7].
Fibroblasts reside in the ECM of healthy tissue to sustain normal tissue homeostasis and are found in an inactive quiescent state under normal physiologic conditions [8]. As a consequence of a disturbance of tissue integrity, such as tissue injury, they become activated and capable of producing transforming growth factor-β (TGF-β) and vascular endothelial growth factor A (VEGF-A) to stimulate wound healing and angiogenesis, respectively [8][9]. Besides, activated fibroblasts are also modulators of the immune response via the secretion of cytokines (e.g., TGF-β and interleukin (IL)-6 [6]) and chemokines (e.g., C-C motif chemokine ligand (CCL) 5, C-X-C motif chemokine ligand (CXCL) 10 [6]), and additionally promote immune surveillance [1][7]. When the process is complete and the activating stimulus is attenuated, the fibroblast activation is reversed to the inactive quiescent state by reprogramming or apoptosis [6]. However, diverse stimuli (reviewed in detail [7]) can cause chronic and irreversible fibroblast activation. This includes inflammatory signals (e.g., IL-1, IL-6, and tumor necrosis factor (TNF)), TGF-β, physical changes in the ECM, contact signals with cancer cells, and DNA damage [7], resulting in a gain of proliferation, secretory phenotype, migration, and ECM production and remodeling [6]. In case of (pre)malignant lesions, persistent accumulation of cancer cells represents an ongoing tissue injury. This initiates a chronic and irreversible hyperactivated fibroblast response towards the cancer cells and eventually the rise of CAFs, with enhanced proliferative properties and self-sustained activation. Additional fibroblast recruitment and proliferation is governed by the release of growth factors (e.g., TGF-β, PDGF, and fibroblast growth factor (FGF) 2) by the cancer cells and infiltrating immune cells [6]. Once CAFs are activated, they can contribute to tumor progression by a broad range of diverse functions.
2. CAFs Contribution to Tumor Progression
2.1. Desmoplasia
The chronic tissue repair response results in the excessive growth of fibrous or connective tissue around an invasive tumor [10]. This process of desmoplasia is achieved through enhanced ECM production and acquired remodeling ability of CAFs [6][10]. As such, desmoplasia causes the tumor to be hard and stiff, leading to a compromised tumor vasculature with blood vessel collapse and hypoxia, thereby promoting formation aggressive tumor clones and inhibition of drug penetration and uptake [7][11]. Furthermore, CAFs and their generated ECM serve as a physical barrier to tumor infiltration by immune cells, and the stiffness of the ECM has shown to enhance cancer cell invasion [6].
2.2. Secretion of Pro-Tumorigenic Modulators
The secretome of CAFs consists of several growth factors and cytokines [8], including connective tissue growth factor (CTGF), epidermal growth factor (EGF), insulin-like growth factor (IGF), hepatocyte growth factor (HGF), basic FGF (bFGF), nerve growth factor (NGF), and IL-6, which promote cancer cell survival as well as its proliferative and invasive behavior. Matrix metalloproteinases (MMPs) released by CAFs remodel the ECM, thus generating permissive tracks to facilitate motility and enhanced invasiveness of cancer cells, eventually boosting metastasis [6][7]. Through the secretion of stromal cell-derived factor 1 (SDF1), next to VEGF-A, CAFs are also able to recruit endothelial progenitor cells into the tumor, thereby stimulating tumor angiogenesis [1][12].
2.3. Generation of a Protumorigenic and Immunosuppressive TME
CAFs are generally considered to promote a protumorigenic and immunosuppressive TME [4][6][7][8][13] directly by releasing a plethora of immunosuppressive as well as immunomodulating and stimulating cytokines (e.g., IL-6, IL-4, IL-8, IL-10, IL-11, IL-17A, TNF, TGF-β, and HGF) and chemokines (e.g., CCL2, CCL5, CCL7, CXCL7, CXCL9, CXCL10, CXCL12, and SDF1), inflammatory factors (e.g., prostaglandin E2 (PGE2)) and immune-modulatory molecules (e.g., human leukocyte antigen (HLA)-G) that can retain suppressive immune subsets and impede proper function of cytotoxic lymphocytes, or indirectly via the remodeling of the ECM, forming a physical barrier for immune cell entry [4][13]. In addition to CAF-derived soluble factors CXCL12, IL-6, and TGF-β, CAFs also inhibit antitumor cytotoxicity of CD8+ T cells through the acquisition of immune checkpoint molecules, such as programmed cell death ligand 1 (PD-L1), PD-L2, and Fas ligand in several tumor types [13][14]. NK cell cytotoxic activity is mainly counteracted by PGE2 and indoleamine 2,3-dioxygenase (IDO) by reducing cytotoxic molecules (granzyme B and perforin) and cytokine release [13]. In summary, CAFs possess the ability to manipulate the immune system, both by excluding and opposing T and NK cell functions via tumor cell downregulation of antigen presentation, elevated expression of surface inhibitory molecules, and secretion of immunosuppressive factors, as well as by maintaining an aberrant inflammatory protumorigenic environment [8][13]. However, CAFs represent a heterogenous population within the TME, with different CAF subsets exerting distinct functions in tumors [4]. This heterogeneity is illustrated by the presence of both myofibroblastic (i.e., myCAF) and non-myofibroblastic (i.e., iCAF) CAF subpopulations, with associated ECM signature and inflammatory phenotype, respectively, in different cancer types (reviewed in detail) [15].
2.4. Mediation of Drug Resistance
CAFs have emerged as key players in mediating drug resistance, and the potential mechanisms to do this are diverse [6][8]. First, CAFs and their generated ECM can function as a physical barrier and thus prevent efficient drug delivery [8]. Moreover, as drug resistance develops over a long period of time, the physical barrier and interaction with the ECM may protect cancer cells from apoptosis while they acquire the mutations needed for cell-intrinsic resistance to therapy [8][16]. Furthermore, CAFs may inhibit the uptake of anticancer drugs as a result of increased intratumoral interstitial fluid pressure, generated by ECM components [6]. Second, the TME might aid cancer cell survival and induce epithelial–mesenchymal transition (EMT) through the enhanced adhesion of cancer cells to the ECM, leading to therapeutic resistance, often described as cell adhesion-mediated drug resistance (CAM-DR) [6][8][17]. As they have been identified as potential drug resistance mediators, CAF-secreted soluble factors TGF-β, IL-6, and HGF may also contribute to therapeutic resistance by binding to cancer cell receptors causing transcriptional changes and by activating non-transcriptional mechanisms including degradation or redistribution of activators of apoptosis (e.g., proapoptotic Bcl-2 member Bim [18] and c-Fas-associated death domain-like IL-1-converting enzyme-like inhibitory protein-long (c-FLIPL)) [19], and increased stability of suppressors of apoptosis and cell cycle regulators (e.g., p27Kip1-associated cell cycle arrest [20]) [6][8]. In addition, upon exposure to conventional chemotherapies, radiotherapy, and targeted agents, CAFs can gain new features with altered functionality and secretion of proteins (e.g., WNT16B [21]) and cytokines (e.g., IL-17A [22]) that drive tumorigenesis, which can ultimately facilitate the development of therapy resistance and contribute to a more aggressive cancer phenotype [7][8].
As CAFs aid tumor development and contribute to therapy resistance [3], CAFs and cancer cells are increasingly viewed as partners in crime. However, the contribution of CAFs is complex and dynamic, with the involvement of various other players of the TME. Given the profound evidence of a generally protumorigenic function of CAFs, there is a considerable heterogeneity and plasticity between different tumor types and divergent context-dependent CAF phenotypes within distinct tumors [6][7][8][23]. Therefore, in this review, we discuss two solid tumor types, i.e., lung cancer and pancreatic cancer, with clear evidence of CAFs contributing to therapy resistance and marked by poor long-term prognosis. We discuss how CAFs contribute to tumor progression, affect their response to clinical therapy and prognosis, and we provide an overview of novel therapies involving CAF-targeting agents for both cancer types.
3. Lung Cancer
Lung cancer remains a devastating disease and the leading cause of cancer-related death worldwide [24], independent of sex [25]. Approximately 84.3% of all lung malignancies are classified as non-small cell lung cancer (NSCLC), for which the predicted 5-year relative survival rate for newly diagnosed cases is 21.0% [25] compared to only 7.0% for small cell lung cancer (SCLC) [26].
Important advancements in the treatment of NSCLC have been achieved over the past two decades due to the introduction of small molecule tyrosine kinase inhibitors and immunotherapy [27]. This is in contrast to SCLC, where immunotherapy shows only minor benefit [28]. However, oncogenic driver mutations are only present in 15–20% of NSCLC patients and PD-L1 tumor proportion score (TPS) ≥ 50% in 28% of advanced NSCLC, thereby limiting the use of these novel treatment strategies [29][30]. As such, surgery, cytotoxic chemotherapy, and radiotherapy remain the backbone for the treatment of both NSCLC [24][31] and SCLC [32]. Nonetheless, the overall cure for NSCLC and SCLC remains low, particularly in metastatic disease [27][28], as the majority of patients have already progressed to a more advanced stage at diagnosis, and due to the occurrence of therapeutic resistance, especially to chemotherapy and targeted therapies [33]. Therefore, further molecular characterization of the tumor landscape is needed, in order to investigate resistance mechanisms and develop novel therapeutic strategies [10].
The TME in lung cancer has been recognized to play a central role in the initiation and progression of primary de novo lung cancer [10], as well as in contributing to therapeutic resistance [34]. Lung tumors harbor distinct subsets of CAFs, each expressing a unique repertoire of collagens and other ECM molecules, demonstrating phenotypic diversity in activated pathways, such as EMT and angiogenesis, that may promote invasiveness and metastasis [35]. Next to resistance resulting directly from CAFs, CAFs are able to promote resistance indirectly by modulating the response to chemotherapy, radiotherapy, targeted therapies, and immunotherapy through paracrine signaling to cancer and immune cells and mutual metabolic reprogramming (illustrated in Figure 1) [36].
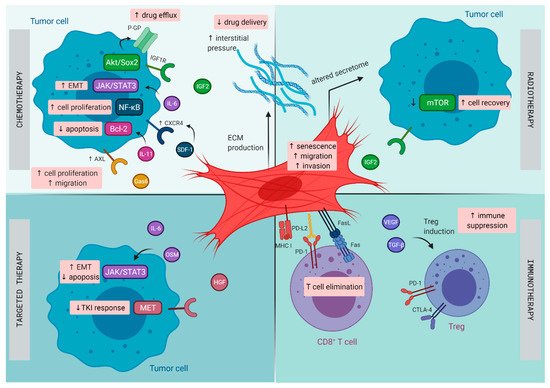
Figure 1. Mechanisms of therapy resistance in lung cancer orchestrated by cancer -associated fibroblasts (CAFs): Schematic illustration on how CAFs diminish the effect of chemotherapy, radiotherapy, targeted therapy, and immunotherapy in lung cancer. CTLA-4, cytotoxic T lymphocyte-associated protein 4; CXCR4, C-X-C chemokine receptor type 4; ECM, extracellular matrix; EMT, epithelial–mesenchymal transition; FasL, Fas ligand; Gas6, growth arrest specific-6 protein; HGF, hepatocyte growth factor; IGF1R, insulin-like growth factor 1 receptor; IGF2, insulin-like growth factor 2; IL, interleukin; MHC I, major histocompatibility complex I; OSM, oncostatin-M; PD-1, programmed cell death 1; PD-L2, programmed cell death ligand 2; P-GP, P-glycoprotein; SDF, stromal cell-derived factor; TGF-β, transforming growth factor-β; TKI, tyrosine kinase inhibitor; Treg, regulatory T cell; VEGF, vascular endothelial growth factor.
4. Pancreatic Cancer
Pancreatic ductal adenocarcinoma (PDAC) is among the deadliest human malignancies. Despite the efforts that have been made, the overall prognosis remains extremely poor, with a 5-year overall survival that still stands below the bar of 10% [37]. Unfortunately, pancreatic cancer is even expected to become the second leading cause of cancer related deaths of the western world within the next decade, due to increasing age and risk factors (e.g., smoking, obesity, and diabetes) [38][39]. These devastating numbers reflect the resistance that is created by the tumor and its environment. Therefore, medical treatment of PDAC remains an ongoing challenge.
PDAC has a unique TME characterized by a dense stromal compartment that can even make up 90% of the whole tumor bulk [40]. This stromal shield is composed of a variety of cell types that are embedded in a dense ECM. CAFs play a central role in the formation of this fibrotic shield and mostly originate from PSCs, a resident stromal cell population of the pancreas [41]. By synthesis and secretion of various ECM components (e.g., collagen, laminin, and fibronectin), CAFs orchestrate the formation of the fibrotic tissue. Due to their presence within the PDAC tumor, along with their secreted factors, they are believed to be the major confounding factor involved in mediating therapeutic resistance (illustrated in Figure 2).
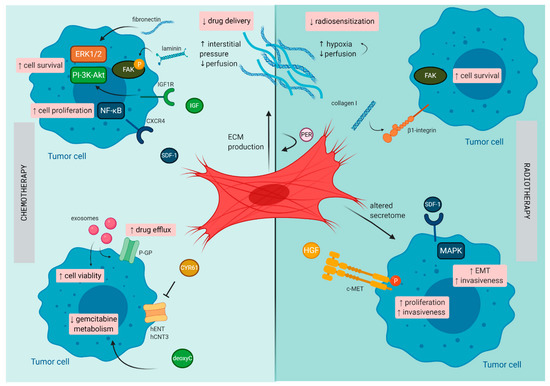
Figure 2. Mechanisms of therapy resistance in pancreatic cancer orchestrated by cancer -associated fibroblasts (CAFs): Schematic illustration on how CAFs diminish the effect of chemotherapy and radiotherapy in pancreatic cancer. c-MET, tyrosine-protein kinase Met CXCR4, C-X-C chemokine receptor type 4; CYR61, cysteine-rich angiogenic inducer 61; deoxyC, deoxycytidine; EMT, epithelial–mesenchymal transition; FAK, focal adhesion kinase; hCNT3, human concentrative nucleoside transporter-3; hENT1, human equilibrative nucleoside transporter-1; HGF, hepatocyte growth factor; IGF, insulin-like growth factor; P, phosphorylated; PER, periostin; P-GP, P-glycoprotein; SDF-1, stromal cell-derived factor-1.
Currently, surgery is the sole curative treatment option for PDAC patients. Unfortunately, most patients present with locally advanced disease or distant metastasis at the time of diagnosis, precluding complete surgical resection of the tumor. Conventional chemotherapy treatment constitutes the standard of care for advanced or metastatic disease. Gemcitabine became the reference chemotherapeutic regimen for advanced PDAC and results in a median overall survival of 6.8 months, or 8.5 months when combined with nab-paclitaxel [42]. Recently, the use of FOLFIRINOX, a combination chemotherapy (oxaliplatin, irinotecan, fluorouracil, and leucovorin) increased the overall survival in these patients to a modest 11.1 months [43]. However, this intensified chemotherapeutic combination is only suitable for patients with a good performance status. Unfortunately, the failure of translating clinical response to chemotherapy into significant survival benefit could be attributed to poor penetration of the administered drug into the tumor and subsequent development of chemoresistance, in which CAFs play a pivotal role [44].
5. CAF-Targeted Treatment
As CAFs display a broad range of mechanisms to stimulate tumorigenesis and drug resistance [8], combined with their genetic stability and relative abundance among stromal cells [36], targeting these cells is becoming an appealing and emerging therapeutic strategy. Several different approaches have been suggested for CAF-targeted anticancer treatment: (1) targeting the biophysical stromal barrier to increase drug delivery, (2) inhibiting CAF-secreted factors that stimulate tumorigenesis and drug resistance, (3) depleting or blocking the ECM components to induce stromal depletion and reduce adhesion-mediated signaling, (4) targeting the CAFs themselves to disable their downstream effects, (5) forcing CAFs to adopt an inactive quiescent phenotype (through the use of molecules such as all-trans retinoic acid (ATRA) or calcipotriol), and (6) modifying and using CAFs as in situ durable reservoir to deliver anticancer drugs (such as oncolytic adenoviruses, TNF-related apoptosis-inducing ligand (TRAIL), or IFN) [8][45]. Here, we will highlight the results of CAF-directed anticancer therapies for lung and pancreatic cancer that have been investigated in clinical studies and those under current clinical investigation (Table 1).
Table 1. Active clinical trials targeting CAF in lung cancer and PDAC.
Lung Cancer | ||||||||||||||||||||
Goal | Compound | Class | ID | Title | Phase | Status | ||||||||||||||
Reduce ECM and IL-1β | Canakinumab | mAB targeting IL-1β | NCT03447769 | Canakinumab as Adjuvant Therapy in Adult Subjects with Stages AJCC/UICC v. 8 II-IIIA and IIIB (T > 5 cm N2) Completely Resected NSCLC | Phase III | Recruiting | ||||||||||||||
Prevent activation of CAFs and block CAF secretome | Erdafitinib | TKI of FGF receptor 1-4 | NCT02699606 | Erdafitinib, A Pan- FGFR Tyrosine Kinase Inhibitor, In Asian Participants with Advanced NSCLC, Urothelial Cancer, Esophageal Cancer Or Cholangiocarcinoma | Phase II | Active, not recruiting | ||||||||||||||
Prevent generation and activation of CAFs | Entinostat | HDAC inhibitor | NCT02437136 | Entinostat with Pembrolizumab in NSCLC with Expansion Cohorts in NSCLC, Melanoma, and Colorectal Cancer | Phase II | Active recruiting | ||||||||||||||
NCT01928576 | Epigenetic Therapy with Azacitidine and Entinostat with Concurrent Nivolumab in Subjects With Metastatic NSCLC | Phase II | Recruiting | |||||||||||||||||
Prevent generation and activation of CAFs | Vorinstat | HDAC inhibitor | NCT02638090 | Pembrolizumab and Vorinostat in Patients with Immune Therapy Naïve and Immune Therapy Pretreated Stage IV NSCLC | Phase I/II | Active recruiting | ||||||||||||||
Prevent generation and activation of CAFs | Mocetinostat | HDAC inhibitor | NCT02805660 | Mocetinostat and Durvalumab in Patients with Advanced Solid Tumors and NSCLC | Phase II | Completed, no results yet | ||||||||||||||
PDAC | ||||||||||||||||||||
Goal | Compound | Class | ID | Title | Phase | Status | ||||||||||||||
Reduce ECM and IL-1β | Canakinumab | mAB targeting IL-1β | NCT04581343 | A Phase 1B Study of Canakinumab, Spartalizumab, Nab-paclitaxel, and Gemcitabine in Metastatic PC Patients (PanCAN-SR1) | Phase Ib | Recruiting | ||||||||||||||
Reduce ECM and TGF-β | Losartan | Angiotensin II receptor antagonist | NCT03563248 | Losartan and Nivolumab in Combination with FOLFIRINOX and SBRT in Localized Pancreatic Cancer | Phase II | Active recruiting | ||||||||||||||
NCT01821729 | Proton w/FOLFIRINOX-Losartan for Pancreatic Cancer | Phase II | Active not recruiting | |||||||||||||||||
Normalize CAFs | ATRA | Vitamin A derivative | NCT04241276 | Phase IIb Randomized Trial of ATRA in a Novel Drug Combination for Pancreatic Cancer (STARPAC2) | Phase IIb | Not yet recruiting | ||||||||||||||
Normalize CAFs | Paricalcitol | Vitamin D analogue | NCT03520790 | Paricalcitol Plus Gemcitabine and Nab-paclitaxel in Metastatic Pancreatic Cancer | Phase I/II | Active, not recruiting | ||||||||||||||
Reduce CAF secretome | Plerixafor | CXCR4 antagonist | NCT04177810 | Plerixafor and Cemiplimab in Metastatic Pancreatic Cancer | Phase II | Recruiting | ||||||||||||||
Reduce CAF secretome | GSK2256098 | FAK inhibitor | NCT02428270 | A Study of GSK2256098 and Trametinib in Advanced Pancreatic Cancer | Phase II | Active, not recruiting | ||||||||||||||
Target FAP+ CAFs | BXCL701 (talabostat) | Small molecule inhibitor of FAP and dipeptidyl peptidases | NCT04123574 | A Pilot Study of BXCL701 in Patients With Pancreatic Cancer | Early phase I | Recruiting | ||||||||||||||
Target FAP+ CAFs | CAR-T targeting nectin4/FAP | CAR-T cell | NCT03932565 | Interventional Therapy Sequential With the Fourth-generation CAR-T Targeting Nectin4/FAP for Malignant Solid Tumors | Phase I | Recruiting | ||||||||||||||
References
- Bremnes, R.M.; Dønnem, T.; Al-Saad, S.; Al-Shibli, K.; Andersen, S.; Sirera, R.; Camps, C.; Marinez, I.; Busund, L.T. The role of tumor stroma in cancer progression and prognosis: Emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 209–217.
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322.
- Santi, A.; Kugeratski, F.G.; Zanivan, S. Cancer Associated Fibroblasts: The Architects of Stroma Remodeling. Proteomics 2018, 18, e1700167.
- Mhaidly, R.; Mechta-Grigoriou, F. Fibroblast heterogeneity in tumor micro-environment: Role in immunosuppression and new therapies. Semin. Immunol. 2020, 48, 101417.
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401.
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598.
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186.
- Öhlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523.
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.L.; Gabbiani, G. The myofibroblast: One function, multiple origins. Am. J. Pathol. 2007, 170, 1807–1816.
- Altorki, N.K.; Markowitz, G.J.; Gao, D.; Port, J.L.; Saxena, A.; Stiles, B.; McGraw, T.; Mittal, V. The lung microenvironment: An important regulator of tumour growth and metastasis. Nat. Rev. Cancer 2019, 19, 9–31.
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; LoBello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; Von Hoff, D.D.; Han, H. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin. Cancer Res. 2015, 21, 3561–3568.
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348.
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835.
- Van Audenaerde, J.R.M.; De Waele, J.; Marcq, E.; Van Loenhout, J.; Lion, E.; Van den Bergh, J.M.J.; Jesenofsky, R.; Masamune, A.; Roeyen, G.; Pauwels, P.; et al. Interleukin-15 stimulates natural killer cell-mediated killing of both human pancreatic cancer and stellate cells. Oncotarget 2017, 8, 56968.
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176.
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674.
- Schmidmaier, R.; Baumann, P. ANTI-ADHESION evolves to a promising therapeutic concept in oncology. Curr. Med. Chem. 2008, 15, 978–990.
- Hazlehurst, L.A.; Argilagos, R.F.; Dalton, W.S. Beta1 integrin mediated adhesion increases Bim protein degradation and contributes to drug resistance in leukaemia cells. Br. J. Haematol. 2007, 136, 269–275.
- Shain, K.H.; Landowski, T.H.; Dalton, W.S. Adhesion-Mediated Intracellular Redistribution of c-Fas-Associated Death Domain-Like IL-1-Converting Enzyme-Like Inhibitory Protein-Long Confers Resistance to CD95-Induced Apoptosis in Hematopoietic Cancer Cell Lines. J. Immunol. 2002, 168, 2544–2553.
- Lwin, T.; Hazlehurst, L.A.; Dessureault, S.; Lai, R.; Bai, W.; Sotomayor, E.; Moscinski, L.C.; Dalton, W.S.; Tao, J. Cell adhesion induces p27Kip1-associated cell-cycle arrest through down-regulation of the SCFSkp2 ubiquitin ligase pathway in mantle-cell and other non-Hodgkin B-cell lymphomas. Blood 2007, 110, 1631–1638.
- Sun, Y.; Campisi, J.; Higano, C.; Beer, T.M.; Porter, P.; Coleman, I.; True, L.; Nelson, P.S. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat. Med. 2012, 18, 1359–1368.
- Lotti, F.; Jarrar, A.M.; Pai, R.K.; Hitomi, M.; Lathia, J.; Mace, A.; Gantt, G.A., Jr.; Sukhdeo, K.; DeVecchio, J.; Vasanji, A.; et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J. Exp. Med. 2013, 210, 2851–2872.
- Zeltz, C.; Primac, I.; Erusappan, P.; Alam, J.; Noel, A.; Gullberg, D. Cancer-associated fibroblasts in desmoplastic tumors: Emerging role of integrins. Semin. Cancer Biol. 2020, 62, 166–181.
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237.
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33.
- Howlader, N.N.A.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; Chen, H.S.; et al. SEER Cancer Statistics Review 1975–2017. Available online: (accessed on 19 January 2021).
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454.
- Rudin, C.M.; Poirier, J.T.; Byers, L.A.; Dive, C.; Dowlati, A.; George, J.; Heymach, J.V.; Johnson, J.E.; Lehman, J.M.; MacPherson, D.; et al. Molecular subtypes of small cell lung cancer: A synthesis of human and mouse model data. Nat. Rev. Cancer 2019, 19, 289–297.
- Mayekar, M.K.; Bivona, T.G. Current Landscape of Targeted Therapy in Lung Cancer. Clin. Pharmacol. Ther. 2017, 102, 757–764.
- Aggarwal, C.; Abreu, D.R.; Felip, E.; Carcereny, E.; Gottfried, M.; Wehler, T.; Ahn, M.-J.; Dolled-Filhart, M.; Zhang, J.; Shentu, Y.; et al. Prevalence of PD-L1 expression in patients with non-small cell lung cancer screened for enrollment in KEYNOTE-001, -010, and -024. Ann. Oncol. 2016, 27, vi363.
- Postmus, P.E.; Kerr, K.M.; Oudkerk, M.; Senan, S.; Waller, D.A.; Vansteenkiste, J.; Escriu, C.; Peters, S. Early and locally advanced non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017, 28, iv1–iv21.
- Früh, M.; De Ruysscher, D.; Popat, S.; Crinò, L.; Peters, S.; Felip, E. Small-cell lung cancer (SCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2013, 24 (Suppl. 6), vi99–vi105.
- Rotow, J.; Bivona, T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658.
- Horvath, L.; Thienpont, B.; Zhao, L.; Wolf, D.; Pircher, A. Overcoming immunotherapy resistance in non-small cell lung cancer (NSCLC)-novel approaches and future outlook. Mol. Cancer 2020, 19, 141.
- Lambrechts, D.; Wauters, E.; Boeckx, B.; Aibar, S.; Nittner, D.; Burton, O.; Bassez, A.; Decaluwé, H.; Pircher, A.; Van den Eynde, K.; et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 2018, 24, 1277–1289.
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol. Cancer 2019, 18, 70.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921.
- Quante, A.S.; Ming, C.; Rottmann, M.; Engel, J.; Boeck, S.; Heinemann, V.; Westphalen, C.B.; Strauch, K. Projections of cancer incidence and cancer-related deaths in Germany by 2020 and 2030. Cancer Med. 2016, 5, 2649–2656.
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A.; Lolkema, M.P.; Buchholz, M.; Olive, K.P.; Gress, T.M.; et al. Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861–868.
- Bynigeri, R.R.; Jakkampudi, A.; Jangala, R.; Subramanyam, C.; Sasikala, M.; Rao, G.V.; Reddy, D.N.; Talukdar, R. Pancreatic stellate cell: Pandora’s box for pancreatic disease biology. World J. Gastroenterol. 2017, 23, 382–405.
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703.
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406.
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157.
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115.